Article Text

Statistics from Altmetric.com
Introduction
In the late 1990s, the discovery that citrullinated proteins are targeted by autoantibodies known as anti-citrullinated protein antibodies (ACPA) revolutionised the study of rheumatoid arthritis (RA). Because of the high specificity of ACPA for RA, investigators began experiments to determine whether ACPA have a direct pathogenetic role in arthritis or whether these antibodies more simply reflect underlying T-cell and B-cell responses without themselves impacting on joint inflammation and damage. The debate on the role of ACPA has been extensive and has now entered a new phase with the publication of correction notes appearing in this issue1 2 of Annals of the Rheumatic (ARD). These notes concern papers that appeared in the journal in 20163 4 and follow a retraction note and correspondence in other journals.5–7
In these notes, the study authors indicate that the monoclonal antibodies used in their experiments on ACPA pathogenicity did not, in fact, react with citrullinated proteins and/or peptides. As such, the putative functional activities of these monoclonals must have resulted from mechanisms other than antibody binding to a citrullinated target. The implications of these corrections and retractions for models of pathogenesis are wide ranging and call into question the nature of ACPA involvement in manifestations of RA. Because of the importance of these issues for the scientific community, in this editorial, we will review the role of ACPA in RA and the salient issues in establishing the pathogenicity of these autoantibodies.
ACPA as biomarkers in RA
ACPA are unique biomarkers for RA, a serious form of arthritis characterised by inflammation, pain and, unless treated effectively, damage of cartilage and bone. As shown in elegant biochemical studies, ACPA target proteins that contain the amino acid citrulline, a post-translational modification (PTM) of the amino acid arginine mediated by peptidyl arginine deiminase enzymes8 9; these antibodies are also called anti-cyclic citrullinated peptide (CCP) antibodies because of their binding to CCP, a synthetic peptide used as a test antigen in immunoassays.8 9 ACPA provide an important criterion in diagnosis and classification.10
While the strong association of ACPA with RA suggests a role in joint inflammation, proof of pathogenicity can be challenging and requires both direct and indirect evidence. Analogous to Koch’s postulates for infectious disease, Witebsky’s postulates for autoimmune disease systemise the experimental evidence needed to demonstrate that a clinical condition is autoimmune and that an autoimmune response (autoantibody or B/T cell) causes pathology.11 For ACPA, the challenge is especially great since ACPA are highly cross-reactive and bind a wide variety of citrullinated proteins; these proteins include citrullinated versions of enolase, fibrinogen, vimentin, collagen and histones, among others.12 13 The relevant target antigen in vivo and its tissue localisation are unknown, although joint tissue in RA shows evidence of citrullination.14
For an autoantibody-mediated disease, pathogenicity is based on the presence of an autoantibody in affected patients; the transfer of disease to an animal by a source of antibodies, including monoclonal antibodies derived from blood or tissue; and induction of disease in an animal model by immunisation with the putative autoantigen. In some situations, an in vitro system can provide a model for a cellular response key to pathogenesis if the relationship to clinical findings is clear. The use of animal models is frequently informative, although it is possible that an autoantibody requires a particular setting (eg, low level of tissue injury or inflammation, the presence of other autoantibodies) for the pathogenicity to be manifest; in this case, the use of wild-type, unmanipulated mice may miss a pathogenic effect.15
Postulated mechanisms for ACPA pathogenicity
For ACPA, two main mechanisms for disease induction or exacerbation have been proposed.16–18 The first mechanism involves the interaction of ACPA and a citrullinated protein to form immune complexes (IC). IC can activate the complement system and induce the release of chemotactic factors such as C3a and C5a to allow recruitment of immune cells to a local site. These cells can subsequently be activated in a fragment crystallisable (Fc)γ-receptor-dependent manner. This activation can lead to the production of cytokines and other pro-inflammatory mediators, culminating in inflammation in general, although arthritis could occur preferentially if the citrullinated proteins are located in the joint or if ACPA production occurs primarily in that local environment. The ability of an ACPA to form an IC may depend on the specificity of the antibody as well as the nature of the citrullinated protein that can be bound in complexed form. IC, including ACPA containing IC, can also bind rheumatoid factor (RF), an autoantibody against IgG which very frequently co-occurs with ACPA; like that of ACPA, the expression of RF can precede the clinical manifestations of the disease and is also important for diagnostic and classification purposes.10 19–21
A more novel mechanism proposed for ACPA pathogenicity relates to the ability of autoantibodies to serve as agonists for a receptor-mediated response. Indeed, these studies, which are the subject of the correction notes, presented intriguing observations indicating that ACPA can directly induce both pain and osteoclastogenesis; these are two cardinal features of RA that can impact on patient symptomatology as well as clinical outcomes.3 4 These studies involved transfer into mice of IgG purified from patient sera as well as monoclonal ACPA to assess pain responses and bone loss; studies on osteoclastogenesis involved in vitro culture systems as well. While both polyclonal and monoclonal antibody preparations produced similar findings in the ARD papers, the authors have meanwhile revealed that the monoclonal antibodies used in these experiments do not bind citrullinated antigens.5
In view of these issues, it is useful to review studies on the pathogenicity of ACPA and determine how studies with monoclonals without apparent ACPA activity could induce pathogenic events in either in vivo or in vitro studies.
The effects of ACPA on inflammation, pain and bone loss
As noted, one potential mechanism by which ACPA can promote events in RA relates to their ability to form IC that can activate cells. Indeed, several studies have demonstrated that the Fc tail of ACPA can activate both Fcγ-receptor expressing cells and the complement system.16 18 Since the expression of citrullinated antigens occurs in the tissue in vivo, including the synovial compartment,14 these studies suggest that ACPA-containing IC can induce inflammation in RA. Importantly, other studies have provided evidence that RF can enhance these effects in vitro, presumably by promoting IC formation via multivalent binding.19–21 These observations link the two characteristic serological findings in RA into one pathogenic pathway and are consistent with clinical findings that arthritis is more severe in patients who express both ACPA and RF.22
While ACPA could drive inflammation by complement system-mediated and Fcγ-receptor-mediated effects, other studies suggest a novel role of ACPA by direct binding to molecules expressed on the surface of cells; these molecules are presumably citrullinated. Indeed, the studies in ARD reported that ACPA can directly induce osteoclastogenesis, thereby contributing to bone loss and joint erosion that are typical for RA. Furthermore, studies by Wigerblad et al in ARD indicated that ACPA can induce pain in an in vivo mouse model by activating osteoclasts to release interleukin 8 and potentially other mediators involved in pain.3 4 The effects observed in these studies appeared to be specific for ACPA since human IgG preparations enriched for ACPA, induced pain behaviour in mice; in contrast, IgG preparations derived from ACPA-negative patients or IgG preparations depleted of ACPA were inactive.
Consistent with the principles embodied in Witebsky’s postulates (which long preceded monoclonal antibody technology), studies with purified IgG from patient blood can provide at least circumstantial evidence for the pathogenicity of a particular antibody specificity. In the ARD publications, the use of monoclonal antibodies derived from patient sources provided perhaps more compelling evidence that the effects on pain and osteoclastogenesis result from direct ACPA binding. In these experiments, murinised monoclonal ACPA, but not monoclonal control antibodies, elicited pain-like behaviour that coincided with the ability to activate osteoclasts. Furthermore, since monomeric Fab fragments were reported to promote osteoclastogenesis,3 23 these effects appeared independent of the Fc region of the antibodies and, hence, from Fcγ-receptor triggering. This last notion is supported by the observation that only monoclonal ACPA displaying a certain epitope-specificity profile could activate osteoclasts, arguing that epitope specificity of the ACPA is crucially involved in the induction of osteoclastogenesis.3 As now known, however, these monoclonal antibodies do not have citrulline specificity.5
In view of the strong associations between ACPA and RA, the findings published in ARD in 2016 attracted great interest as they could explain two prominent features of RA-joint pain and bone erosion in a disease-specific manner. As such, these observations entailed a paradigm shift in thinking about disease pathogenesis by suggesting that certain disease manifestations in RA could arise directly from RA-specific autoantibodies and do not entail IC formation. In this conceptualisation of RA, ACPA would resemble autoantibodies that directly activate or functionally perturb cells such as autoantibodies found in Graves’ disease which bind the thyrotropin receptor stimulating the production of thyroxine and triiodothyronine.
In ascribing pathogenicity to an autoantibody in any disease, a relevant consideration is plausibility (ie, does clinical evidence support the proposed mechanism?). In RA, a number of clinical situations impact on the plausibility that ACPA are pathogenic either by IC formation or agonistic activity. The first concerns the apparent lack of transfer of disease during pregnancy despite the placental transfer of autoantibodies. The expression of ACPA in asymptomatic individuals during the phase of pre-disease autoimmunity is another clinical scenario to consider. Another circumstance relates to the continued expression of ACPA following successful treatment of RA that can diminish pain and halt bone loss. While some reduction in ACPA levels occurs with therapy, this reduction is limited.24 In these various settings, why does not the presence of ACPA lead to either pain or bone loss?
Re-evaluation of data on ACPA pathogenicity
While the concept that ACPA are agonistic agents is intriguing, the new findings ask for a critical re-interpretation of this possibility. Notably, the authors who described the monoclonal ACPA with osteoclastogenic and pain-inducing potential notified the community that, in reassessing the original data, the antibodies in question showed ‘no measurable affinity for the tested citrullinated peptides’ as detailed in a recent correction7 and retraction note5; in other words, the monoclonal antibodies studied were not ACPA. This observation is in line with crystallographic studies of some of these monoclonal antibodies that could not confirm the binding of citrullinated peptides.25 While there is a major unknown— the identity of the antigens that led to a positive signal on synovial tissue and/or osteoclasts—these antibodies had, nevertheless, in vivo and in vitro effects. As also acknowledged by the authors in the two correction notes published in this issue of ARD, it is now clear that ‘the functional results reported for these monoclonal antibodies cannot be attributed to reactivity against citrullinated proteins and/or peptides, but are due to other yet unknown mechanisms’.1 2
In studies on pathogenicity, determination of antibody specificity is crucial and requires appropriate assays to detect binding at relevant concentrations. For ACPA, several different antigens can be used for immunochemical determinations. These include citrullinated proteins and citrullinated peptides especially when presented in large arrays in which a multitude of sequences can be assessed. Other considerations in assessing antibody specificity relate to the detection method (eg, ELISA, surface plasmon resonance, western blotting) that may, in turn, be influenced by technical factors such as salt concentrations and even the nature of the buffer.
For monoclonal antibodies, polyreactivity and cross-reactivity (ie, ability to bind to multiple different antigens or the ability to bind to multiple citrullinated and/or carbamylated/acetylated antigens) may be a complicating factor. Polyreactivity may result from an aberrant ‘pattern of antigen-driven selection’ in the setting of autoimmunity. In autoimmune disease, the repertoires and signalling thresholds for both T and B cells may be abnormal, skewing the binding properties of induced antibodies. In addition to disturbances in B-cell and T-cell function, antigen drive by multiple different antigens may lead to patterns of reactivity unlike those of a conventional antibody response where antigen binding is more specific and has higher avidity.26–28 In this regard, since the nature of self-antigens in vivo is often not known, extrapolating from in vitro assays with purified antigens to that which occurs in vivo can be uncertain especially when an antibody can bind to structurally similar molecules in a cross-reactive manner.
Issues in experimental design and performance
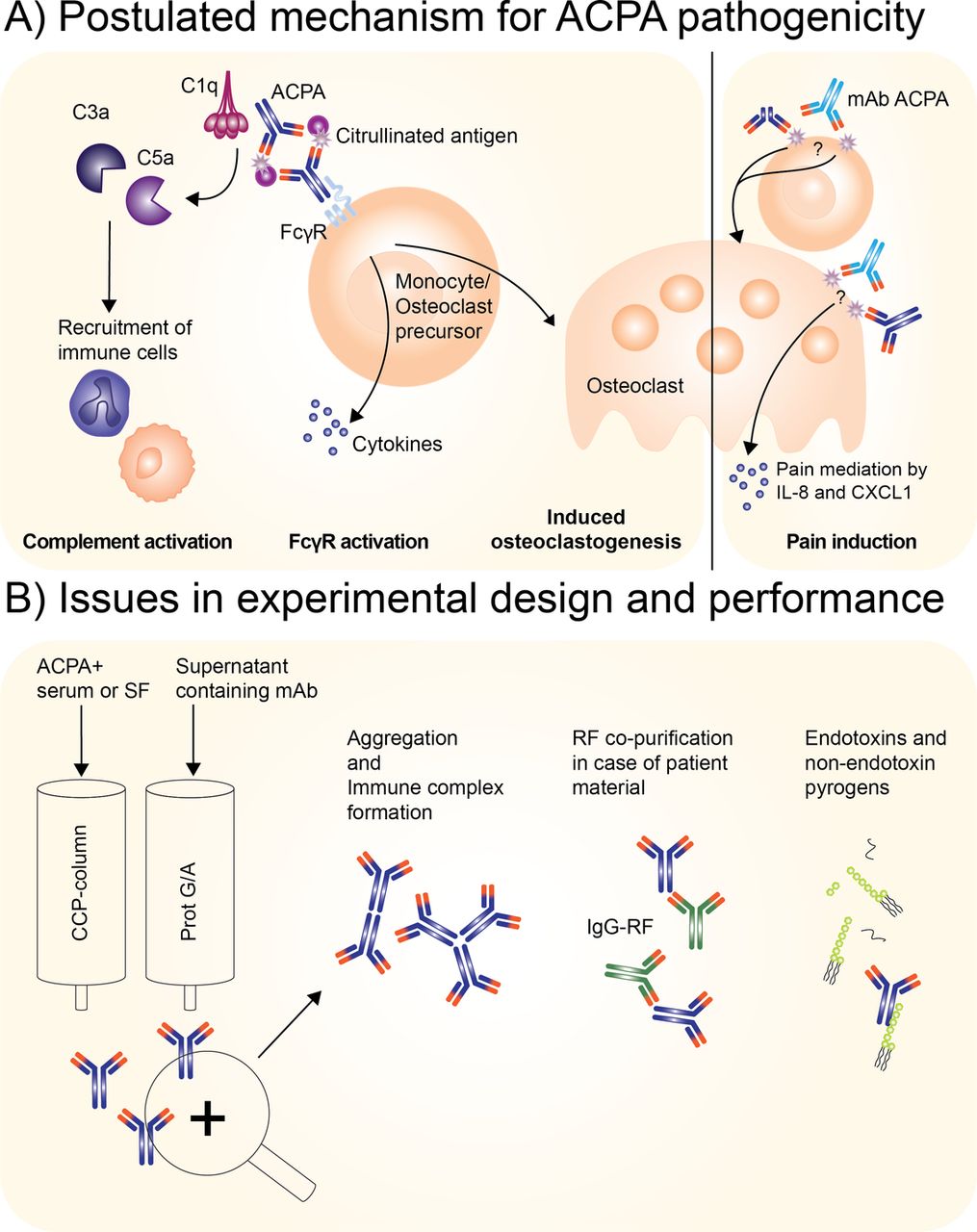
Given these considerations as well as notices of correction and retraction, current data indicate that the biological effects observed with the monoclonal antibodies appear unrelated to the recognition of citrulline as a critical determinant of specificity.5 6 25 Hence, other mechanisms, unrelated to the recognition of citrullinated antigens, must have contributed to the results obtained (figure 1). Whether this situation also pertains to the effects of the affinity-purified polyclonal ACPA containing Ig preparations is more difficult to know. Importantly, however, the authors of the 2016 ARD papers on pain induction and osteoclastogenesis state that ‘also these results have to be interpreted with caution waiting for additional mechanistic studies’.1 2 With polyclonal antibodies, it is important to absorb reactivity with citrullinated versus control antigens to show that, in the first case, the biological effects are eliminated while, in the control condition, they are retained. Still, the binding of antibodies to affinity columns provides presumptive evidence of specificity, but does not prove it. Regardless, since the identification and molecular characterisation of any putative citrullinated receptor have not yet been accomplished, caution is needed in the interpretation of results with the polyclonal preparations as well as monoclonal antibodies. Of note, the avidity of ACPA appears to be relatively low, potentially limiting their ability to directly activate cellular receptors.29 30
{kind=link}
Operational and postulated principles in defining the pathogenic activity of polyclonal or monoclonal antibodies. (A) Postulated mechanism for ACPA pathogenicity either by polyclonal ACPA and/or monoclonal ACPA. Left side: confirmed autoantibody-mediated contributions to arthritis facilitated by complement system activation and Fcγ receptor activation;16 – 18 right side: postulated contribution of ACPA to RA as now under debate indicated by correction notes published in this issue 1 2 to papers published in ARD 3 4 as well as a preceding retraction and correspondence in other journals. 5 – 7 The questions marks indicate the re-evaluation of the pathogenic activity of the monoclonal ACPA, as it is reassessed to be not specific for citrullination. (B) Potential issues occurring during antibody isolation of either ACPA from patient sera or supernatant containing recombinant monoclonal antibodies. Factors that can occur during purification: aggregation, immune complex formation, co-purification of RF in case of ACPA+ serum or synovial fluid and lastly introduction of endotoxin and non-endotoxin pyrogens. Light blue antibody: monoclonal ACPA; dark blue: ACPA from patient IgG fraction enriched for ACPA; asterisk: citrullinated protein; green antibody: RF IgG. ACPA, anti-citrullinated protein antibody; RA, rheumatoid arthritis; RF, rheumatoid factor.
While the relevant specificity of both the polyclonal and monoclonal antibody preparations remains an open question, the finding on the induction of pain and bone loss is nevertheless very interesting and would benefit from reasonable and plausible explanations to move the field forward. In retrospect, several possibilities could be at play. For example, it is possible that the monoclonal and polyclonal antibody preparations contained contaminants that by themselves might have caused biological effects. Myeloid cells, for example, are highly sensitive to endotoxin and non-endotoxin pyrogens; this sensitivity can differ from that of conventional endotoxin-detection kits, such as the Limulus Amebocyte Lysate test. Endotoxin and other pyrogens can be co-purified during protein isolation due to hydrophobic interactions, for example.31 32 Such events can occur variably among antibody preparations, explaining why some monoclonal antibodies or their respective purified Fab preparations activate osteoclasts, whereas others do not.
Another contributing factor relates to the immunochemical properties of an antibody molecule. Among the monoclonal antibodies that could induce osteoclastogenesis and joint pain, D10 and B02 were reported to show ‘polyreactivity’ towards at least two antigens out of three analysed (ie, to double-stranded DNA, insulin and/or lipopolysaccharide (LPS). This activity contrasts with that of at least one of the monoclonal antibodies which did not appear to elicit these effects (RA1276:01:C07). Likewise, because of aggregation that can occur during affinity purification or during the isolation of monoclonal antibodies, certain monoclonal antibody preparations can act as ICs to activate FcγR-positive cells; the production of heavy chain dimers can lead to the same effect. In this situation, batch-to-batch and IgG-to-IgG variations can occur.33–36 Such variability could also explain why some (monoclonal) antibody preparations induce effects in cellular systems, whereas others do not. Similarly, the presence of IgG-RF in IgG preparations purified from serum could also lead to confounding results.
It is important to consider that ACPA might be particularly prone to aggregation and interactions with pyrogens since they display some unique features including the presence of negatively charged, N-linked glycans in the variable domain.37 38 Also in this case, the composition of the N-linked glycans present on recombinantly produced antibodies may vary substantially from batch-to-batch and/or between different antibodies when not controlled for and hence could impact on biological outcomes.39–41 The experience of one of us (RT) with isolated ACPA, monoclonal ACPA, as well as control antibody preparations is consistent with this possibility since we also obtained inconsistent and variable effects in similar studies addressing the osteoclastogenic effects of ACPA (unpublished data). In this context, variable effects seem to be present in the published experiments as well since, in a recent study, one of four monoclonal ACPA induced osteoclastogenesis slightly, two had no effect and the fourth inhibited osteoclastogenesis; as such, when antibodies are present together as may happen during disease, there may be no net effect, with one effect on osteoclast activation counteracted by another.42
Similarly, it has been reported that the osteoclastogenic potential of ACPA occurs only with cells from wild-type mice, but not with cells derived from Fcγ- or FcγRIII knockout mice,43 a notion that seems to conflict with the observation that Fab domains may be sufficient to mediate the effects described. In this respect, it might be relevant to also re-evaluate the reported osteoclastogenic potential of Fab fragment isolated against citrullinated vimentin.23 The observations obtained with cells from FcγR knockout mice are, however, in line with the findings that (heat)-aggregated IgGs display a strong osteoclastogenic potential; this potential may be more pronounced for aggregated IgG preparations enriched for ACPA, perhaps related to the reduced sialylation of the N-linked Fc glycans which enhances FcγR binding.44
Implications for the future
The publication of retraction and correction notes is always unfortunate because of the effects on investigators especially when unforeseen technical issues have affected experiments. Beyond effects on investigators, however, retractions and corrections, especially if delayed, also negatively impact the scientific community as a whole since investigators who were attracted by reported findings may have invested time and effort to explore these observations, leading them in a perhaps wrong direction or deterring them from pursuing other potential mechanisms. Correcting the literature will also take time especially as novel ideas often receive prominence in reviews and other articles.
While disappointing and regrettable, these circumstances nevertheless provide an opportunity to rethink concepts as well as improve experimental designs. Given publication of several studies on agonist activities of ACPA, these considerations do not exclude the possibility that ACPA (or even a related specificity such as anti-malondialdehyde [monoclonal] antibodies as recently proposed)45 can directly activate osteoclasts to induce bone resorption and pain; providing new evidence in support of this phenomenon is crucial, especially to explain the activity of polyclonal antibody preparations.
Adding to the complexity of the ACPA system is the extensive cross-reactivity of these autoantibodies towards different citrullinated antigens as well as other PTMs expressed by proteins.42 46 47 As these different modifications are recognised with varying avidities, attributing biological effects of monoclonal or polyclonal antibodies to the recognition of citrulline alone is difficult and it can be debated whether citrulline specificity and/or the ‘Peptidyl Arginine Deiminase-Citrulline’ pathway should be considered the predominant or indeed only auto-reactive phenomenon. In this respect, it is important to note that recent epidemiological data may not support the postulated connection between smoking and formation of citrullinated proteins and a breach of tolerance to citrullinated antigens48; thus, smoking may be associated with the concurrent presence of multiple autoantibodies in RA rather than with ACPA per se.49–51 Ultimately, it will be essential to delineate fully the autoantibody repertoire in RA and to clone, express and to define at the molecular level the possible cellular receptors targeted by ACPA, assessing the relevant PTM. In view of the experience with ACPA, we would suggest the following operational principles in defining the pathogenic activity of either polyclonal or monoclonal antibodies (Box 1).
Considerations for defining the pathogenic activity of either polyclonal or monoclonal antibodies
Assess antibody activity using well-validated assays especially those that are clinically useful.
Validate assays sufficiently to ensure that they detect specific antibody binding and not non-specific interactions.
Assure antibody specificity by direct binding and inhibition studies of polyclonal and monoclonal antibodies.
For affinity purification, consider use of different antigens (eg, different citrullinated proteins) for the affinity matrix and assess both bound and unbound fractions for antibody binding and functional activity.
Remove aggregates by ultracentrifugation or size-exclusion chromatography.
Assess for contamination by endotoxin and related material.
For in vitro studies, perform dose–response curves and assess activity at relevant antibody concentrations.
For in vivo studies, assess activity in different strains of mice including knockout mice to define mechanisms.
For in vivo studies, consider the use of models in which subclinical arthritis is induced.
For both in vivo and in vitro studies, consider the effects of rheumatoid factor.
In case of agonistic antibodies binding to cellular receptors, clone the receptor from the target cells for expression in reporter cells to mimic agonistic activity.
Conclusions
The recent information on the binding properties of monoclonal antibodies once considered to be ACPA will require a period of reflection and re-evaluation of prior results on the potential pathogenicity of ACPA in RA. Nevertheless, the data presented in a number of papers suggest that autoantibodies can mediate events in RA by diverse mechanisms, including IC formation and, possibly, direct agonist activity. Clearly, as the field moves forward, the claims that ACPA can activate cells implicated in pathogenesis should receive more extensive and rigorous experimental support before they can be accepted. Nonetheless, as our understanding of the unique features of ACPA and other autoantibodies grows, these mechanisms will undoubtedly undergo further investigation. In the quest to understand the possible contribution of ACPA to disease pathogenesis, these efforts will provide new and stimulating insights into the pathways contributing to RA pathogenesis and hopefully allow new approaches to diagnosis, staging and treatment.
Acknowledgments
We thank Dr Hans Ulrich Scherer for critically reading the manuscript and Ms Lise Hafkenscheid for drawing of figure 1.
References
Footnotes
Handling editor Josef S Smolen
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Commissioned; internally peer reviewed.