Article Text

Abstract
Objective Osteoarthritis (OA) appears to be associated with various metabolic disorders, but the potential contribution of amino acid metabolism to OA pathogenesis has not been clearly elucidated. Here, we explored whether alterations in the amino acid metabolism of chondrocytes could regulate OA pathogenesis.
Methods Expression profiles of amino acid metabolism-regulating genes in primary-culture passage 0 mouse chondrocytes were examined by microarray analysis, and selected genes were further characterised in mouse OA chondrocytes and OA cartilage of human and mouse models. Experimental OA in mice was induced by destabilisation of the medial meniscus (DMM) or intra-articular (IA) injection of adenoviruses expressing catabolic regulators. The functional consequences of arginase II (Arg-II) were examined in Arg2−/− mice and those subjected to IA injection of an adenovirus encoding Arg-II (Ad-Arg-II).
Results The gene encoding Arg-II, an arginine-metabolising enzyme, was specifically upregulated in chondrocytes under various pathological conditions and in OA cartilage from human patients with OA and various mouse models. Adenovirus-mediated overexpression of Arg-II in mouse joint tissues caused OA pathogenesis, whereas genetic ablation of Arg2 in mice (Arg2 −/−) abolished all manifestations of DMM-induced OA. Mechanistically, Arg-II appears to cause OA cartilage destruction at least partly by upregulating the expression of matrix-degrading enzymes (matrix metalloproteinase 3 [MMP3] and MMP13) in chondrocytes via the nuclear factor (NF)-κB pathway.
Conclusions Our results indicate that Arg-II is a crucial regulator of OA pathogenesis in mice. Although chondrocytes of human and mouse do not identically, but similarly, respond to Arg-II, our results suggest that Arg-II could be a therapeutic target of OA pathogenesis.
- chondrocytes
- cartilage
- osteoarthritis
- arginase II
- matrix metalloproteinases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Abnormalities of cellular arginine-metabolising enzymes, arginases, have been associated with multiple diseases, but its potential contribution to osteoarthritis pathogenesis has not been clearly elucidated.
What does this study add?
We demonstrate that arginase II (Arg-II), which is upregulated in osteoarthritic cartilage tissue, regulates osteoarthritis pathogenesis in mice by modulating the expression levels of matrix-degrading enzymes in chondrocytes.
How might this impact on clinical practice or future developments?
Arg-II could be a therapeutic target in efforts to combat osteoarthritis pathogenesis.
Introduction
Osteoarthritis (OA) carries a large socioeconomic cost, but we currently lack an effective disease-modifying therapy.1 This whole-joint disease involves cartilage destruction, synovitis, osteophyte formation and subchondral bone remodelling.2 3 Among these manifestations, cartilage destruction, which is a hallmark of OA, is caused by the actions of matrix-degrading enzymes,4 such as matrix metalloproteinase 3 (MMP3), MMP13, and ADAMTS5.5–7 In chondrocytes, these enzymes are regulated by various extracellular catabolic regulators, including proinflammatory cytokines.8 We previously identified various cellular catabolic mediators in chondrocytes, including hypoxia-inducible factor (HIF)-2α,9 the zinc importer ZIP8,10 oestrogen-related receptor γ (ERRγ)11 and BATF (basic leucine zipper transcription factor, ATF-like),12 all of which play catabolic functions by upregulating matrix-degrading enzymes.9–12
OA appears to be associated with metabolic disorders,13–15 and alterations in amino acid metabolism are associated with OA pathogenesis.16 For instance, the inducible nitric oxide (NO) synthase (iNOS)-mediated production of NO from arginine in chondrocytes is associated with OA pathogenesis.17 Amino acids serve as precursors for various metabolites and bioactive molecules, in addition to acting as essential building blocks for protein synthesis. Therefore, it is reasonable to hypothesise that alterations of amino acid metabolism could be associated with OA pathogenesis. Our group preliminarily examined this hypothesis by microarray analysis, and found that the gene encoding the arginine-metabolising enzyme, arginase II (Arg-II), was markedly increased in chondrocytes treated with proinflammatory cytokines or overexpressing cellular catabolic mediators of OA, such as HIF-2α or ZIP8. Because the role of Arg-II in OA has not previously been investigated, we herein explored the possible functions and underlying molecular mechanisms of Arg-II in OA pathogenesis. We found that Arg-II is specifically upregulated in human and mouse OA cartilage. Our gain-of-function (adenovirus-mediated overexpression) and loss-of-function (Arg2 −/− mice) approaches indicated that Arg-II acts as a novel catabolic regulator of OA pathogenesis at least partly by upregulating the matrix-degrading enzymes, MMP3 and MMP13, in chondrocytes.
Materials and methods
Human OA cartilage was sourced from individuals undergoing arthroplasty (online supplementary table S1).9–12 Arg2−/− mice18 (Jackson Laboratory) and wild-type (WT) littermates were used for the experimental OA studies. The experimental procedures are detailed in the online supplementary materials and methods and online supplementary table S2. These include the induction of experimental OA; histological analyses; primary cell culture; microscopy; microarray; transcription factor (TF) array; reverse transcription-PCR (RT-PCR) and quantitative RT-PCR (qRT-PCR); western blotting; and assays of arginase activity, senescence, autophagy, reactive oxygen species (ROS) production, mitochondrial dysfunction, NO production and nuclear factor (NF)-κB reporter gene. Statistical comparisons of two independent groups were made using the Shapiro-Wilk test for normality, Levene’s test for homogeneity of variance and the two-tailed independent t-test. Multiple comparisons were performed using the Shapiro-Wilk test, Levene’s test and one-way analysis of variance (ANOVA) with post - hoc Bonferroni test. Data based on ordinal grading systems were analysed by the non-parametric Mann-Whitney U test. The n numbers indicate the number of independent primary cultures or mice per group. The p values are indicated in the figures, and the error bars represent SEM for parametric data and the calculated 95% confidence invervals (CIs) for non-parametric data. Additional information on the statistical tests reflected in the individual figures is presented in online online supplementary tables S3 and S4.
Supplemental material
Results
Arg-II is upregulated in OA chondrocytes
The expression profiles of genes involved in amino acid metabolism were examined by microarray analysis of primary-culture passage 0 (P0) mouse chondrocytes treated with stimuli that trigger OA pathogenesis, namely interleukin (IL)-1β8 or the adenovirus-mediated overexpression of HIF-2α (Ad-HIF-2α)9 or the zinc importer ZIP8 (Ad-ZIP8).10Among the examined genes, the mRNA levels of those encoding arginine-metabolising enzymes were markedly increased in the stimulated chondrocytes (figure 1A; online supplementary supplementary table S5). Among the examined arginine-metabolising enzymes, the mRNA levels of Arg-II and iNOS (NOS2) were the most prominently increased by IL-1β, Ad-HIF-2α or Ad-ZIP8 (figure 1B). While the role of iNOS-mediated NO production in OA pathogenesis has been extensively studied,17 the role of Arg-II had not previously been investigated in this context. Accordingly, we examined the functions of Arg-II in OA pathogenesis. RT-PCR (online supplementary figure S1A–D) and qRT-PCR (figure 1C) analyses confirmed that Arg-II mRNA levels were significantly increased in chondrocytes treated with IL-1β or tumour necrosis factor (TNF)-α, or infected with Ad-HIF-2α or Ad-ZIP8. Immunostaining of cartilage sections indicated that Arg-II protein levels were markedly elevated in OA-affected damaged regions of human cartilage compared with undamaged areas from the same patient (figure 1D) and in mouse models of OA cartilage caused by destabilisation of the medial meniscus (DMM) surgery or intra-articular (IA) injection of Ad-HIF-2α or Ad-ZIP89 10(figure 1E).

Arg-II is upregulated in OA chondrocytes. (A and B) Microarray analysis of primary-culture P0 mouse chondrocytes stimulated with IL-1β, Ad-HIF-2α or Ad-ZIP8 (n=4). Heat map of genes involved in amino acid metabolism (A) and arginine metabolism (B). (C) Relative Arg-II mRNA levels in chondrocytes stimulated as indicated. The data (mean±SEM; one-way analysis of variance with Bonferroni test) were obtained from 12 (IL-1β), 5 (TNF-α) and 10 (Ad-HIF-2α and Ad-ZIP8) independent primary cultures of mouse chondrocytes. (D) Representative images of Alcian blue staining and Arg-II immunostaining of human OA cartilage (n=7 patients). (E) Representative images of Safranin-O and immunostaining of Arg-II, HIF-2α and ZIP8 in sham-operated or DMM-operated mouse cartilage (n=10 mice/group) or cartilage of mice intra-articular-injected with Ad-C (n=6 mice), Ad-HIF-2α (n=10 mice) or Ad-ZIP8 (n=10 mice). Scale bar: 50 µm. Ad-HIF-2α, adenovirus-mediated overexpression of HIF-2α; Ad-ZIP8, adenovirus-mediated overexpression zinc importer; Arg-II, arginase II; DMM, destabilisation of the medial meniscus; HIF, hypoxia-inducible factor; IL, interleukin; MOI, multiplicity of infection; OA, osteoarthritis; P0, passage 0; TNF-α, tumour necrosis factor-α.
Arg-II overexpression in mouse joint tissues causes OA
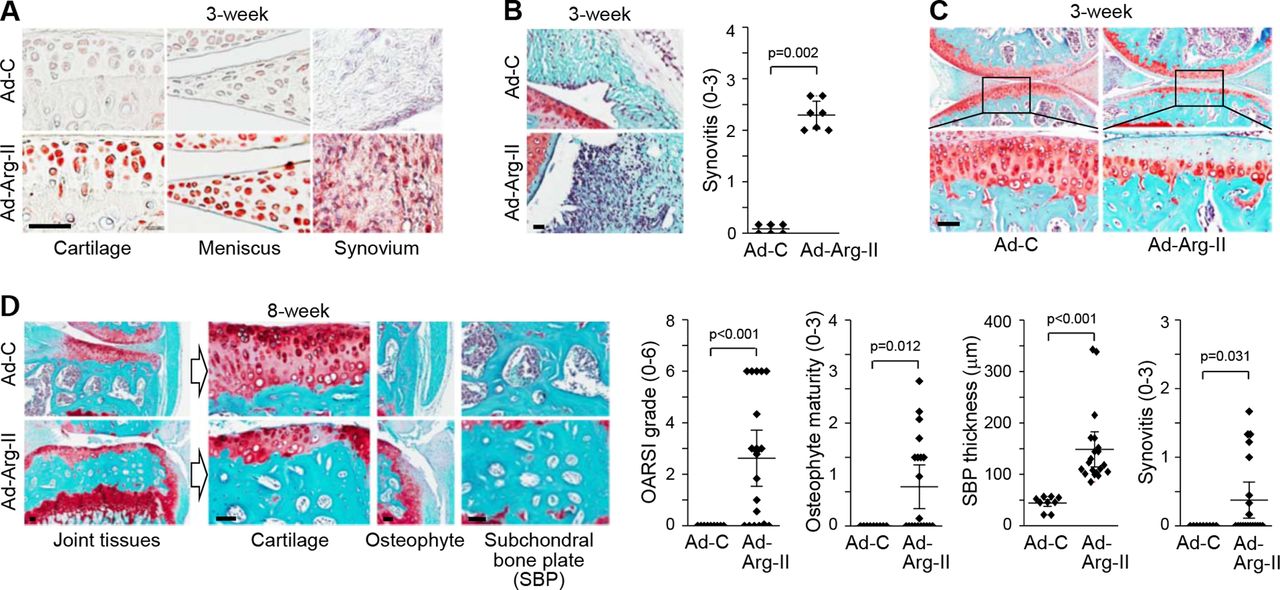
The in vivo functions of Arg-II were examined by its overexpression in knee-joint tissues via IA injection of Ad-Arg-II in 12-week-old male mice. We had previously demonstrated that an adenovirus system could effectively deliver genes to cartilage and other joint tissues.9–12 Here, we confirmed that Arg-II was effectively expressed in the cartilage, meniscus and synovium of joint tissues (figure 2A). At 3 weeks post-IA injection, Arg-II overexpression was associated with synovitis (figure 2B) and loss of glycosaminoglycans in articular cartilage without apparent osteophyte development or subchondral bone remodelling (figure 2C). At 8 weeks post-IA injection, joint tissues showed severe cartilage erosion, osteophyte development and thickening of the subchondral bone plate (suggestive of sclerosis) (figure 2D). These results collectively suggest that Arg-II functions as a catabolic regulator in mouse models of OA pathogenesis.

Arg-II overexpression in mouse joint tissues causes OA. (A−C) WT mice were IA-injected with Ad-C (n=6 mice) or Ad-Arg-II (n=7 mice) and sacrificed at 3 weeks postinjection. Representative images of Arg-II immunostaining (A), Safranin-O/haematoxylin staining and scoring of synovitis (B), and Safranin-O staining of cartilage sections (C). (D) WT mice were IA-injected with Ad-C (n=9 mice) or Ad-Arg-II (n=20 mice) and sacrificed at 8 weeks postinjection. Representative images of Safranin-O staining of joint sections are presented. Values (B and D) are presented as means±95% CI; Mann-Whitney U test (OARSI grade, osteophyte maturity and synovitis) and two-tailed t-test (SBP thickness). Scale bar: 50 µm. Arg-II, arginase II; IA, intra-articular; OA, osteoarthritis; OARSI, Osteoarthritis Research Society International; WT, wild-type.
As studies have shown that synovitis contributes to OA pathogenesis19 and FLS (fibroblast-like synoviocytes) regulate synovitis by producing various OA-associated catabolic factors,20 we examined whether Arg-II in mouse FLS modulates the expression of matrix-degrading enzymes (MMPs and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS)), cytokines (IL-1β, IL-6 and TNF-α), and chemokines and their receptors (CC chemokine ligand (CCLs) and CC chemokine receptor (CCRs)). We found that Arg-II overexpression in FLS did not modulate the expression of the examined catabolic factors (online supplementary figure S2). However, it remains possible that Arg-II overexpression in other cell types of synovial tissues in mice IA injected with Ad-Arg-II may contribute to OA pathogenesis.
Arg2−/− mice exhibit protection from experimental OA
We further examined the functions of Arg-II in OA pathogenesis using Arg2−/− mice. These mice are viable18 and exhibited normal development of cartilage and other skeletal elements, including the skeletal size of E18.5 embryos, the growth plate structure in 2-week-old metatarsal bone and the structure of 5-month-old cartilage tissues (online supplementary figure S3A-D). However, compared with WT littermates, Arg2−/− mice exhibited significant reductions in all manifestations of DMM-induced OA, including cartilage erosion, osteophyte formation and subchondral bone plate thickening (figure 3A,B). The DMM-induced upregulation of Arg-II was completely abrogated in Arg2−/− mice (figure 3A). Consistent with the observed inhibition of cartilage destruction, the DMM-induced upregulations of MMP3 and MMP13 (figure 3C) and productions of the aggrecan neoepitopes, NITEGE and DIPEN21 (figure 3D), which were seen in the cartilage of WT mice, were markedly reduced in Arg2−/− mice. These results suggest that Arg-II is necessary for OA pathogenesis in mouse models.
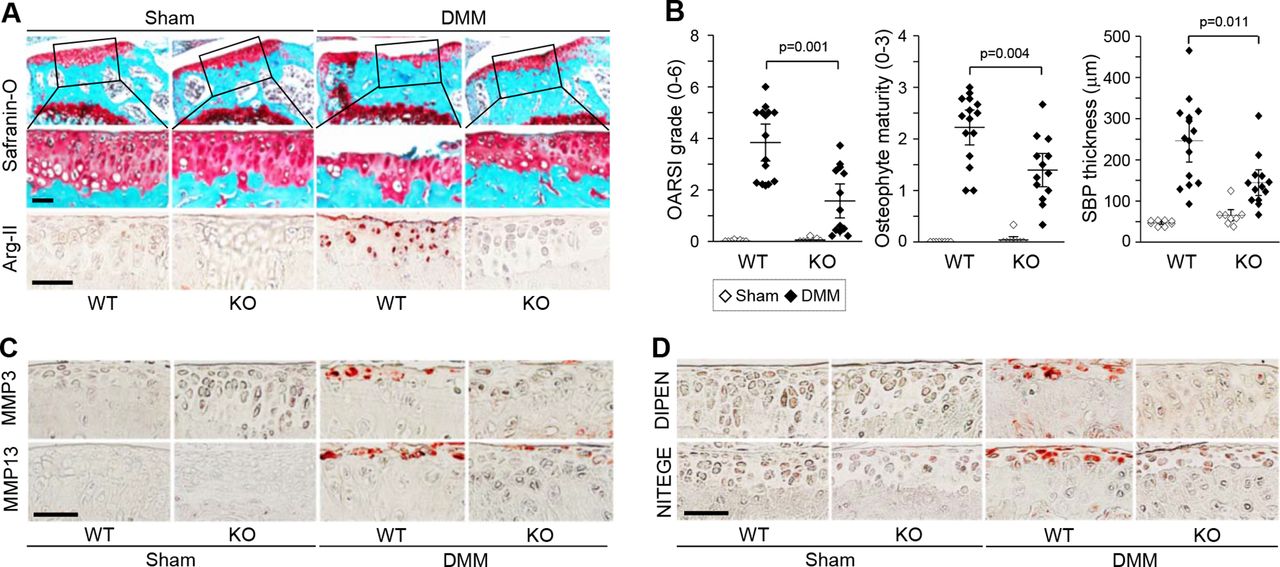
Arg2−/− mice exhibit protection from post-traumatic OA. Arg2−/− mice and WT littermates were subjected to sham operation (n=8 mice/group) or DMM surgery (n=15 mice for WT and n=13 mice for knock-out (KO)) and sacrificed at 8 weeks postoperation. Representative images of Safranin-O staining and Arg-II immunostaining in joint sections (A), quantitation of OA parameters (B), and representative immunostaining images of MMP3 and MMP13 (C) and the aggrecan neoepitopes, DIPEN and NITEGE (D), in cartilage sections. Values are presented as means±95% CI; Mann-Whitney U test (OARSI grade and osteophyte maturity) and two-tailed t-test (SBP thickness). Scale bar: 50 µm. DMM, destabilisation of the medial meniscus; MMP, matrix metalloproteinase; OA, osteoarthritis; OARSI, Osteoarthritis Research Society International; WT, wild-type.
Arg-II upregulates MMP3 and MMP13 in mouse articular chondrocytes
To elucidate the mechanisms underlying Arg-II regulation of OA cartilage destruction, we examined the impact of Arg-II on the expression levels of matrix-degrading enzymes in chondrocytes. As genetic studies in mice have demonstrated that MMP3, MMP13 and ADAMTS5 play crucial roles in OA cartilage destruction,5–7 we examined these and other factors in our system. Arg-II overexpression in primary-culture chondrocytes markedly elevated the mRNA levels (figure 4A,B) and extracellular secreted protein levels of MMP3 and MMP13 (figure 4C). However, Arg-II overexpression in chondrocytes did not modulate ADAMTS5 at the mRNA or protein levels (figure 4A–C). In addition, Arg-II overexpression did not modulate the mRNA levels of SOX9, type II collagen or aggrecan (figure 4A). The overexpression of Arg-II in cartilage via IA injection of Ad-Arg-II caused the upregulation of MMP3 and MMP13 (figure 4D), confirming that Arg-II causes cartilage destruction by upregulating MMP3 and MMP13. Although ADAMTS5 expression was not modulated by Arg-II, the productions of the DIPEN and NITEGE neoepitopes were markedly elevated in Ad-Arg-II-injected cartilage tissue (figure 4E).

Arg-II upregulates matrix-degrading enzymes in mouse chondrocytes. (A−C) Primary cultures of mouse chondrocytes were infected with Ad-C (800 MOI) or the indicated MOI of Ad-Arg-II. Presented are representative RT-PCR results (A), qRT-PCR results (B) and western blot results for cellular Arg-II and secreted MMP3, MMP13 and ADAMTS5 (C). The data were obtained from nine (A and B) and five (C) independent primary cultures of chondrocytes. Values are presented as means±SEM (one-way analysis of variance with Bonferroni test). (D and E) WT mice were IA-injected with Ad-C (n=9 mice) or Ad-Arg-II (n=20 mice) and sacrificed at 8 weeks postinjection. Representative images of Safranin-O staining and immunostaining of MMP3 and MMP13 (D) and detection of the Arg-II and DIPEN and NITEGE neoepitopes (E). Scale bar: 50 µm. Arg-II, arginase II; ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; Ad-Arg-II, adenovirus encoding Arg-II; IA, intra-articular; MOI, multiplicity of infection; MMP, matrix metalloproteinase; qRT-PCR, quantitative RT-PCR; RT-PCR, reverse transcription-PCR; WT, wild-type.
NF-κB mediates the Arg-II-induced upregulation of MMP3 and MMP13
Arg-II in chondrocytes was localised in the mitochondria (figure 5A), which is consistent with previous reports.22–25 Stimulation of chondrocytes with IL-1β or Ad-Arg-II increased the expression and enzymatic activity of Arg-II (figure 5B). Because both enzymatic and non-enzymatic functions of Arg-II regulate various cellular processes,25–27 we examined whether arginase enzymatic activity is necessary for the Arg-II-induced upregulation of MMPs. Whereas WT-Arg-II increased the mRNA levels of MMP3 and MMP13, △-Arg-II (H160F),26 which lacks arginase activity (figure 5D), did not (figure 5C). This indicates that arginase activity is necessary for the Arg-II-induced expression of MMP3 and MMP13 in chondrocytes.

Arg-II upregulates MMPs via the NF-κB pathway in mouse chondrocytes. (A) Representative confocal microscopic images of Mitotracker and Arg-II staining in chondrocytes (n=4). (B) Arginase activity in chondrocytes treated as indicated (n=4). (C and D) mRNA levels (C; n=8) and arginase activity (D; n=5) in chondrocytes transfected with 1 µg of EV, WT-Arg2 or △-Arg2. (E) TF activity in chondrocytes infected with Ad-Arg-II. (F) NF-κB reporter gene activity in chondrocytes transfected with EV or vectors encoding WT-Arg2, Δ-Arg2 or p65, or stimulated with IL-1β (n=9). (G) mRNA levels in chondrocytes infected with Ad-Arg-II and exposed to SC-514 (n=6). The n indicates the number of independent primary cultures of chondrocytes. Values are presented as means±SEM (one-way analysis of variance with Bonferroni test). AP-1, activator protein 1; Arg-II, arginase II; Ad-Arg-II, EV, empty vector; GR, glucocorticoid receptor; IL-1β, interleukin-1β; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB; PPAR, peroxisome proliferator-activated receptor; TF, transcription factor.
TF array analysis revealed that Arg-II overexpression in chondrocytes caused more than twofold increases in the activities of E2F, NF-κB, PPAR (peroxisome proliferator-activated receptor), SMAD, AP-1 (activator protein 1) and GR (glucocorticoid receptor) (figure 5E; online supplementary figure S4A). Inhibition of NF-κB with SC-51428 markedly abrogated the Arg-II-induced upregulations of MMP3 and MMP13, whereas inhibition or knockdown of other TFs exhibited no marked effect (online supplementary figure S4B). Consistently, WT-Arg-II significantly increased NF-κB reporter gene activity (figure 5F). △-Arg-II also activated NF-κB, although to a significantly smaller degree than seen with WT-Arg-II (figure 5F). Therefore, both enzymatic and non-enzymatic activities of Arg-II contribute to NF-κB activation, whereas MMP expression is regulated by the enzymatic activity of Arg-II. Consistent with this, inhibition of NF-κB partially but significantly inhibited the Arg-II-induced upregulation of MMPs (figure 5G). Moreover, while NF-κB inhibition completely abrogated the IL-1β-induced upregulation of MMP3 and MMP13, Arg2−/− chondrocytes exhibited significant but incomplete inhibition of the IL-1β-induced upregulations of MMP3 and MMP13 (online supplementary figure S5A-E). Our results collectively indicate that Arg-II activates NF-κB, and that this significantly contributes to, but does not fully account for, the Arg-II-induced expression of MMP3 and MMP13 in chondrocytes.
Because Arg-II enzymatic activity is necessary for MMP expression, we examined whether the enzymatic products of Arg-II, ornithine and urea, could mimic the effects of Arg-II overexpression in chondrocytes. Similar to the effects of Arg-II overexpression on the expression of matrix-degrading enzymes, exogenous ornithine, but not urea, caused upregulation of MMP3 and MMP13 and activation of NF-κB (online supplementary figure S6A-D). Our results suggest that stimulation of Arg-II and subsequent production of ornithine is associated with NF-κB activation and MMP expression in chondrocytes.
Arg-II modulates NO production and chondrocyte senescence
Arg-II regulates oxidative stress, mitochondrial dysfunction and autophagy in various cell types.26 27 29 However, Arg-II-overexpressing or Arg2−/− chondrocytes did not exhibit any change in ROS production (figure 6A), mitochondrial membrane potential (figure 6B), or the autophagy of serum-starved and nutrient-starved chondrocytes (figure 6C). Because Arg-II competes with NOS for arginine,22–25 we examined whether Arg-II could modulate NO production. IL-1β-induced NO production was significantly reduced by the overexpression of Arg-II, but a large amount of NO was still produced in these cells (figure 6D). Consistently, IL-1β caused more NO production in Arg2−/− cells compared with WT chondrocytes (figure 6D). However, the observed inhibition of DMM-induced OA in Arg2−/− mice (figure 3) indicates that this enhancement of NO production in Arg2−/− chondrocytes is not associated with the ability of Arg-II to regulate OA pathogenesis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Arg-II modulates NO production and senescence in mouse chondrocytes. (A) ROS production in chondrocytes infected with Ad-C or Ad-Arg-II (n=4). Tert-butyl hydroperoxide (TBHP) was used as a positive control. (B) JC-1 assay in chondrocytes infected with Ad-Arg-II or in Arg2−/− chondrocytes (n=4). Valinomycin (10 µM) was used as a positive control. (C) Autophagy was induced by serum/nutrient starvation in chondrocytes infected with Ad-C or Ad-Arg-II. Autophagy markers were detected by western blotting (n=5). (D) NO production in IL-1β-stimulated chondrocytes infected with Ad-C or Ad-Arg-II (Ad-A) or in WT or Arg2−/− chondrocytes (left n=13 and right n=4). (E) SA-β-gal staining in passage 0 (P0) chondrocytes infected with Ad-C or Ad-Arg-II for 72 hours or in p6 WT and Arg2−/− chondrocytes. Quantitation of SA-β-gal-positive cells (n=9) and p16INK4A mRNA levels (n=9). (F) Schematic illustration of the functions of Arg-II in OA pathogenesis. The n numbers indicate the numbers of independent primary cultures of chondrocytes. Values are presented as means±SEM one-way analysis of variance with Bonferroni test (D, left) and two-tailed t-test (D, right, and E). Ad-Arg-II, adenovirus encoding Arg-II; Arg-II, arginase II; DAPI, 4',6-diamidino-2-phenylindole; DHE, dihydroethidium; DMM, destabilisation of the medial meniscus; FITC-A, fluorescein isothiocynate (FITC)-A; HIF, hypoxia-inducible factor; IL-1β, interleukin-1β; NO, nitric oxide; OA, osteoarthritis; ROS, reactive oxygen species; SA-β-gal, senescence-associated β-galactosidase; TBHP, tert-butyl hydroperoxide; ZIP8, zinc importer.
Finally, we examined whether Arg-II modulates chondrocyte senescence, as this is a well-known regulatory mechanism of OA pathology30 31 and Arg-II is known to regulate senescence in various cell types.26 32 However, Arg-II overexpression in P0 WT chondrocytes did not cause senescence, as demonstrated by senescence-associated β-galactosidase (SA-β-gal) staining and detection of p16INK4A mRNA levels (figure 6E). Serial-passage culture of WT chondrocytes to P6, which causes de-differentiation,33 resulted in positive SA-β-gal staining and increased mRNA expression of p16INK4A; in contrast, these parameters were significantly reduced in Arg2−/− chondrocytes subjected to serial passage (figure 6E). Therefore, although Arg-II overexpression in P0 chondrocytes does not cause senescence, the lack of Arg-II inhibits the cellular senescence caused by serial passage culture.
Human relevance of Arg-II regulation in OA pathogenesis
To support the human relevance of our findings, we examined the potential roles of Arg-II in human chondrocytes. We found that cartilage tissue of all seven patients examined herein exhibited elevation of Arg-II protein levels (figure 1D). To examine the role of Arg-II in human chondrocytes, we used two different lots (donors) of P2 human normal chondrocytes (Cell Applications) and P1 human OA chondrocytes sourced from three individuals undergoing arthroplasty (online supplementary table 1). Our experiments revealed that Arg-II overexpression increased the mRNA levels of MMP3 without effects on the MMP13 mRNA levels both in human normal chondrocytes (online supplementary figure S7A, B) and OA chondrocytes (online supplementary figure S7E, F). Our results indicate that Arg-II plays similar, although not identical, functions in mouse and human chondrocytes in the context of MMP expression. Lastly, we examined the potential role of Arg-II in the senescence of human chondrocytes. As seen in mouse chondrocytes, overexpression of Arg-II in P2 human normal chondrocytes or P1 human OA chondrocytes did not cause cellular senescence, as determined by SA-β-gal staining (online supplementary figure S7C, G). However, unlike the mouse results, serial-passage culture of human normal and OA chondrocytes to P6 did not cause cellular senescence, and knockdown of Arg-II in P6 human chondrocytes also did not affect SA-β-gal staining (online supplementary figure S7D, H). Our results are consistent with previous report that more than 10 passages are required for senescence of human chondrocytes.34
Discussion
Abnormalities of cellular arginase have been associated with multiple diseases, including those of the cardiovascular and central nervous systems.23 Here, we demonstrate that Arg-II regulates OA pathogenesis in mice by modulating the expression levels of MMP3 and MMP13 in chondrocytes. Arg-II is upregulated in mouse chondrocytes subjected to mechanical stress (DMM), extracellular catabolic regulators (proinflammatory cytokines) and cellular catabolic mediators (HIF-2α and ZIP8). The upregulation of Arg-II is necessary for OA pathogenesis in mice, as demonstrated by our gain-of-function and loss-of-function approaches. Our results collectively indicate that Arg-II is a crucial catabolic regulator of OA pathogenesis in mice, as summarised in figure 6F.
We found that Arg-II, but not Arg-I, is upregulated in chondrocytes under our examined pathological conditions. Arg-I and Arg-II are encoded by separate genes; they both convert arginine to urea and ornithine,22–24 but knock-out (KO) studies in mice have demonstrated that they have isoform-specific functions. Arg1−/− mice die before postnatal day 14 with severe hyperammonia,35 whereas Arg2−/− mice are viable18 and exhibit protection from obesity-associated pancreatic cancer36 and high-fat diet-induced hepatic steatosis.37 Arg-I and Arg-II also differ in their cellular localisations and expression mechanisms. Arg-I is cytoplasmic and its expression is inducible by extracellular stimuli, whereas Arg-II is located in the mitochondria and constitutively expressed in many cell types.22–24 Here, we found that Arg-II is specifically upregulated in chondrocytes stimulated with proinflammatory cytokines or by the overexpression of HIF-2α and ZIP8, whereas Arg-I is barely detectable in these chondrocytes. Notably, Arg-I is the most strongly regulated mechanosensitive gene induced immediately after the induction of OA by DMM surgery and on ex vivo cartilage injury.38 39 Therefore, whereas Arg-II is stimulated by extracellular catabolic regulators and cellular catabolic mediators, Arg-I is likely to be highly active in mechanically damaged tissues of the joint.
The NF-κB-mediated upregulations of MMP3 and MMP13 in chondrocytes appear to be associated with the regulation of Arg-II during cartilage destruction. Our results suggest that complex signalling pathways govern Arg-II-induced NF-κB activation and the downstream induction of MMPs. Both enzymatic and non-enzymatic functions of Arg-II caused NF-κB activation, whereas the enzymatic activity of Arg-II was solely responsible for the inductions of MMP3 and MMP13. Arg-II-induced MMP expression was only partially blocked by the inhibition of NF-κB, whereas the IL-1β-induced upregulation of MMPs was completely abrogated. Therefore, it is likely that complicated and yet-unknown signalling mechanisms regulate Arg-II-induced MMP expression in chondrocytes. Nevertheless, we found that ornithine, an enzymatic product of Arg-II, causes NF-κB activation and MMP induction. Although we did not elucidate the mechanisms underlying these effects, we speculate that ornithine or its metabolites (polyamines such as putrescine, spermidine and spermine) mediate the effects of Arg-II on the expression levels of matrix-degrading enzymes. Among these enzymes, MMP3, MMP13 and ADAMTS5 play crucial roles in OA cartilage destruction.5–7 The activity of ADAMTS5, which regulates aggrecan degradation, is for the most part controlled post-translationally.40 However, we found that Arg-II does not modulate the mRNA or protein levels of ADAMTS5 in chondrocytes. Therefore, the inhibition of the DMM-induced production of aggrecan neoepitopes in Arg2−/− mice is likely to be mediated by other aggrecanases. Additionally, MMP3 is essential for collagenase-induced OA5 but not for cartilage destruction in DMM-induced OA.41 Therefore, it is likely that the NF-κB-mediated upregulation of MMP13 plays an important role in the Arg-II-mediated cartilage destruction seen in experimental mouse models of OA.
Chondrocyte senescence is believed to contribute to OA pathology, with OA chondrocytes exhibiting a variety of senescence-associated phenotypes.31 In various cell types, Arg-II has been shown to interact with ribosomal S6 kinase and mitochondrial p66Shc to promote cell senescence, apoptosis and inflammation under pathological conditions.26 32 Moreover, a recent study indicated that Arg-II deficiency extends the lifespan in mice.42 However, no previous report has addressed the functions of Arg-II in regulating chondrocyte senescence. We observed here that Arg-II deficiency significantly inhibited SA-β-gal staining in serial-passage-cultured P6 chondrocytes. This suggests that Arg-II regulates subculture-induced chondrocyte senescence, which might be associated with OA pathogenesis.31 However, ectopic expression of Arg-II in P0 chondrocytes did not alter SA-β-gal staining, indicating that simple overexpression of Arg-II does not cause chondrocyte senescence. Therefore, the detailed mechanisms through which Arg-II regulates chondrocyte senescence remain to be further elucidated.
The human relevance of the ability of Arg-II to regulate OA pathogenesis was supported by the upregulation of Arg-II in human OA cartilage. Additionally, overexpression of Arg-II similarly, although not identically, regulated the expression of matrix-degrading enzymes in mouse and human chondrocytes. Although Arg-II functions in the senescence of human chondrocytes are not clearly determined in this study, our results collectively suggest that our findings are relevant to human OA and that Arg-II could be a therapeutic target in efforts to combat OA.
References
Footnotes
W-SC and J-IY contributed equally.
Handling editor Professor Josef S Smolen
Contributors W-SC and J-IY: study design, data acquisition, data analysis, data interpretation, manuscript preparation and manuscript approval. WK, H-EK, S-KK, YW and Y-OS: data acquisition, data analysis and manuscript approval. C-HC provided and evaluated the human joint samples. J-SC takes responsibility for the integrity of this work.
Funding This work was supported by grants from the National Research Foundation of Korea (2016R1A3B1906090 and 2016R1A5A1007318) and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (HI16C0287 and HI14C3484). W-SC and J-IY were supported by grants from the National Research Foundation of Korea (2017R1A6A3A11034719 and 2015H1A2A1034404, respectively).
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval The Institutional Review Board of Wonkwang University Hospital approved the use of human materials. All animal experiments were approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.