Article Text

Abstract
Objective To assess the efficacy, safety, pharmacokinetics and pharmacodynamics of the anti-interleukin (IL)-1α/β dual variable domain immunoglobulin lutikizumab (ABT-981) in erosive hand osteoarthritis (HOA).
Methods Patients with ≥1 erosive and ≥3 tender and/or swollen hand joints were randomised to placebo or lutikizumab 200 mg subcutaneously every 2 weeks for 24 weeks. The primary endpoint was change in Australian/Canadian Osteoarthritis Hand Index (AUSCAN) pain subdomain score from baseline to 16 weeks. At baseline and week 26, subjects had bilateral hand radiographs and MRI of the hand with the greatest number of baseline tender and/or swollen joints. Continuous endpoints were assessed using analysis of covariance models, with treatment and country as main factors and baseline measurements as covariates.
Results Of 132 randomised subjects, 1 received no study drug and 110 completed the study (placebo, 61/67 (91%); lutikizumab, 49/64 (77%)). AUSCAN pain was not different among subjects treated with lutikizumab versus placebo at week 16 (least squares mean difference, 1.5 (95% CI –1.9 to 5.0)). Other clinical and imaging endpoints were not different between lutikizumab and placebo. Lutikizumab significantly decreased serum high-sensitivity C reactive protein levels, IL-1α and IL-1β levels, and blood neutrophils. Lutikizumab pharmacokinetics were consistent with phase I studies and not affected by antidrug antibodies. Injection site reactions and neutropaenia were more common in the lutikizumab group; discontinuations because of adverse events occurred more frequently with lutikizumab (4/64) versus placebo (1/67).
Conclusion Despite adequate blockade of IL-1, lutikizumab did not improve pain or imaging outcomes in erosive HOA compared with placebo.
- DMOADs (biologic)
- hand osteoarthritis
- interleukin-1
- inflammation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
What is already known about this subject?
Preclinical studies of human osteoarthritis (OA) and mouse models suggested that interleukin-1α (IL-1α) and IL-1β were potential mediators of synovitis, cartilage damage, and bone loss in patients with erosive hand OA (HOA). Lutikizumab is a dual variable domain immunoglobulin (DVD-Ig) that binds and neutralises both IL-1α and IL-1β.
What does this study add?
In this phase 2a, randomised, double-blind study in patients with erosive HOA, treatment with lutikizumab, versus placebo, did not result in significantly better improvements in hand joint pain, despite adequate neutralization of IL-1α and IL-1β.
Besides hand pain, other symptomatic, functional, and structural efficacy endpoints were not significantly different for erosive HOA patients treated with lutikizumab versus placebo.
Injection site reactions, neutropenia, and discontinuations because of AEs were more frequent with lutikizumab compared with placebo.
How might this impact on clinical practice?
Targeting IL-1 may not be effective for the treatment of erosive HOA.
Introduction
Hand osteoarthritis (HOA) is highly prevalent, especially among women and the elderly.1 2 Erosive HOA, characterised by pain, swelling, interphalangeal joint inflammation and central erosions,3 4 can result in substantial disability.5–7 Treatments are limited8 to topical and oral non-steroidal anti-inflammatory drugs (NSAIDs) to relieve pain9; there are no disease-modifying OA drugs that prevent structural damage.8 9 Antitumour necrosis factor (anti-TNF) therapies in patients with erosive HOA failed to demonstrate significant pain relief but did slow structural progression of inflamed joints.10 11 A randomised, placebo-controlled study (Hydroxychloroquine Effectiveness in Reducing symptoms of hand Osteoarthritis (HERO)) showed no significant symptomatic or radiographic efficacy for hydroxychloroquine in patients with severe HOA.12 The efficacy of intra-articular steroid injections has been equivocal in patients with HOA.13
Inflammatory cytokines such as interleukin-1 (IL-1) may be involved in the pathogenesis of OA.14 15 IL-1α and IL-1β bind to the IL-1 type 1 receptor (IL-1R1), leading to the production of proinflammatory molecules, proteases and other mediators,16 17 which result in joint pain, inflammation and cartilage destruction.15 IL-1 is also present in the bone,18 where it plays an important role in bone resorption.18–20 In a rabbit model, production of transfected exogenous IL-1 receptor antagonist (IL-1Ra) reduced the severity of induced knee OA lesions.21 In clinical trials, an IL-1Ra drug (anakinra)22 and an antibody (AMG 108) that blocks the IL-1R123 were generally well tolerated, but did not significantly improve symptoms in patients with knee OA. However, in contrast to knee OA,24 erosive HOA is associated with an IL-1β gene polymorphism,25 and in a case series (n=3) anakinra markedly improved pain in patients with erosive HOA,26 implying that inhibition of IL-1 might be beneficial.
Lutikizumab (ABT-981), a novel human dual variable domain immunoglobulin (DVD-Ig), simultaneously binds and inhibits IL-1α and IL-1β without interfering with other human IL-1 family members including IL-1Ra.27 In a mouse model, a mouse anti-IL-1α/β DVD-Ig reduced OA progression28 and increased the threshold for evoked pain29 more than inhibition of either IL-1α or IL-1β alone. In a phase I multiple ascending-dose study in subjects with knee OA, lutikizumab was generally well tolerated and associated with expected pharmacodynamic effects, including reductions in neutrophils, high-sensitivity C reactive protein (hsCRP) and reductions in markers of synovitis (matrix metalloproteinase (MMP)-degraded collagen type III (C3M), MMP-generated fragment of CRP (CRPM) and MMP-degraded collagen type 1 (C1M)).30 The objective of the current study was to determine the clinical and structural efficacy, safety, pharmacokinetics and pharmacodynamics of lutikizumab in patients with erosive HOA.
Methods
Study design
The design of this phase IIa, randomised, double-blind, placebo-controlled, parallel-group study (NCT02384538; EudraCT 2014-001096-31) in patients with erosive HOA (online supplementary figure 1) was based on the recommendations of an Osteoarthritis Research Society International (OARSI) task force.31 All subjects provided informed consent. After a screening and washout period of approximately 45 days, subjects were randomised 1:1 to placebo or lutikizumab 200 mg subcutaneously every 2 weeks for 24 weeks (13 injections) using stratification by country (online supplementary methods).
Supplemental material
Supplemental material
Subjects
Adult patients 35–80 years diagnosed with HOA and fulfilling the American College of Rheumatology criteria32 were eligible. Key inclusion criteria included active inflammation (ie, ≥3 tender and/or swollen interphalangeal joints), subject-rated hand pain ≥6 (11-point Numeric Rating Scale (NRS-11), 0–10), and radiographic evidence of ≥1 interphalangeal joint defined by Verbruggen et al 33 as erosive (E; subchondral plate eroded) or erosive/remodelling (E/R; new irregular sclerotic subchondral plates with intervening space in a part of the joint). Radiographs were centrally scored by CP for eligibility. Subjects had to have discontinued use of analgesics, NSAIDs and nutraceuticals (online supplementary methods).
Key exclusion criteria included previous anti-IL-1 treatment; corticosteroid use within 1 month before screening; immunosuppressive therapy within 1 month (biologic) or 3 months (conventional) or 5 half-lives, whichever was longer, before beginning the study drug; colchicine within 1 month before beginning the study drug; diagnosis of fibromyalgia, inflammatory arthritis, gout, pseudogout, secondary OA, psoriatic arthritis or psoriasis; active or latent, untreated tuberculosis; and evidence of premalignant dysplasia or history of malignancy within 5 years, except successfully treated non-metastatic cutaneous squamous cell, basal cell carcinoma or localised carcinoma in situ of the cervix. Patients with a positive hepatitis A, B or C virus and HIV test were excluded with evidence of active infection (vs past infection) or history of HIV.
Efficacy
The primary endpoint was change from baseline to 16 weeks in the Australian/Canadian Osteoarthritis Hand Index score (AUSCAN V.3.1; NRS-11, range 0–50 pain subdomain score) (online supplementary methods).34 A key secondary endpoint was change from baseline to 26 weeks in AUSCAN function subdomain score (NRS-11, range 0–90).34 Key exploratory endpoints included change from baseline to 26 weeks in swollen and tender joint counts (each totalling 0–30 for metacarpophalangeal, proximal interphalangeal, distal interphalangeal, first interphalangeal and first carpometacarpal joints in both hands), structural bone and joint damage by radiography, and synovitis, erosive damage, cartilage space loss and bone marrow lesions by MRI (online supplementary figure 1, online supplementary methods). To assess structural damage, at baseline and week 26, subjects had radiography of both hands and MRI of the hand with the largest number of tender and/or swollen joints at baseline (index hand). Radiographs were scored according to a modified Verbruggen-Veys method,35 the OARSI atlas36 and a modified Kellgren-Lawrence Scale.37 38 MRIs were scored using a modified Outcome Measures in Rheumatology/Hand Osteoarthritis MRI Scoring system (OMERACT/HOAMRIS).39
Central readers, independent of AbbVie, separately scored (online supplementary table 1) the radiographs (IKH and FK) and the MRIs (CP and Yan Chen, Spire Sciences). Scores of the pairs of readers were averaged for both radiographic and MRI analyses. However, if an adjudicator score was available (online supplementary table 2), it was substituted for both radiographic and MRI analyses instead of an average score. Discrepancies between readers in radiographic change scores above a predefined threshold were adjudicated by a third independent reviewer (Gust Verbruggen, Ghent University Hospital). The top 10% of discrepancies in scores between readers for each feature of the OMERACT/HOAMRIS readings were adjudicated by consensus review to identify and correct potential input errors. All radiographic and MRI scoring were blinded to examination chronology and subject and clinical characteristics.
Supplemental material
Supplemental material
Rescue medication
Subjects could take acetaminophen (maximum, 3000 mg/day) as rescue medication during the washout period through week 16 or acetaminophen (maximum, 3000 mg/day) and/or ibuprofen (maximum, 1200 mg/day) during weeks 16–26 for breakthrough hand pain. Rescue medication was requested to be stopped 48 hours before clinical outcome assessments at baseline and at each biweekly clinic visit.
Pharmacokinetics
Blood samples were taken at baseline and throughout the 26 weeks to assess lutikizumab levels and antidrug antibody responses to lutikizumab (online supplementary methods).
Target engagement and pharmacodynamics
To measure target engagement, IL-1α and IL-1β serum levels were measured at baseline and weeks 2 and 4. Pharmacodynamic measures included absolute neutrophil counts (ANCs) at baseline and every 2 weeks thereafter, serum hsCRP measured at baseline and weeks 2, 4, 6, 8, 12, 16, 20 and 26, and serum C1M, C3M, CRPM, hyaluronic acid, N-propeptide of collagen IIA (PIIANP), and C-terminal telopeptide fragments of type I collagen (CTX-I) and urine CTX-II (corrected for urine creatinine) measured at baseline and weeks 4, 16 and 26 (see online supplementary methods for assay methods).
Safety
Adverse events (AEs), vital signs, physical examinations and laboratory data were assessed throughout the study (online supplementary methods).
Statistical analyses
Sample size was determined based on the primary efficacy variable, change from baseline in AUSCAN pain subdomain score. In published HOA and erosive HOA studies, the mean baseline AUSCAN pain score ranged from 23 to 33,10 40–42 and the SD of change from baseline in these scores ranged from 6.7 to 14.10 40 42 43 The sample size per group (n=60) was estimated using 10% inflation to account for possible dropouts, with 80% power at α=0.05, assuming SD=10 and based on a two-sided, two-sample t-test for detecting a difference of 5.4 (20%) between lutikizumab and placebo.
Efficacy was analysed in a modified intent-to-treat population comprising randomised subjects who received ≥1 dose of the study drug (online supplementary methods). Continuous efficacy endpoints were assessed using analysis of covariance models, with treatment and country as main factors and baseline measurements as covariates. Last observation carried forward (LOCF) imputation was used for the primary endpoint; observed cases were used for other endpoints. Sensitivity analysis using a mixed model that assumed randomly missing data revealed only modest differences from LOCF, with no impact on the conclusions (data not shown). The safety analysis set included subjects who received ≥1 dose of the study drug per treatment assignment.
Results
Subjects
In this study, 558 patients were screened, 132 randomised and 131 treated, from 29 April 2015 to 13 July 2016. The most common reasons for exclusion were absence of erosions, hand pain <6 and laboratory abnormalities (online supplementary table 3). Of treated subjects, 61 of 67 (91%) who received placebo and 49 of 64 (77%) who received lutikizumab completed the study (online supplementary figure 2). In the placebo group, 1 of 67 (1%) and in the lutikizumab group 4 of 64 (6%) subjects discontinued from the study because of AEs. Demographic and baseline characteristics were well matched between treatment groups (table 1).
Supplemental material
Supplemental material
Demographics and baseline disease characteristics
Efficacy
Pain, function and joint symptoms
The primary efficacy endpoint, least squares (LS) mean (95% CI) change from baseline in AUSCAN pain at 16 weeks, was similar for placebo (−10.7 (–15.4 to –6.0)) and lutikizumab (−9.2 (−13.8 to –4.6)) (LS mean difference (95% CI), 1.5 (–1.9 to 5.0); LOCF; figure 1A). At 16 weeks, the LS mean (95% CI) change from baseline in AUSCAN function was −17.2 (–24.9 to –9.4) for placebo and −14.6 (–22.1 to –7.1) for lutikizumab (LS mean difference (95% CI), 2.5 (−3.2 to 8.3); LOCF; figure 1B). Changes from baseline at other time points in AUSCAN pain (figure 1A) and function (figure 1B) were similar in subjects treated with lutikizumab compared with placebo. The LS mean change from baseline in tender and swollen joint counts at week 26 was similar between placebo and lutikizumab (table 2). Other efficacy outcomes (pain, stiffness, grip strength and patient-reported outcomes) were also not different between the placebo and lutikizumab groups (online supplementary table 4).
Supplemental material

LS mean change from baseline in AUSCAN pain over time (A) and LS mean change from baseline in AUSCAN function over time (B). Last observation carried forward imputation was used for analysis when values were missing. All comparisons of lutikizumab versus placebo were not significant using an analysis of covariance adjusted for treatment group and country as factors, and including baseline value as a covariate. AUSCAN, Australian/Canadian Osteoarthritis Hand Index; LS, least squares.
Imaging
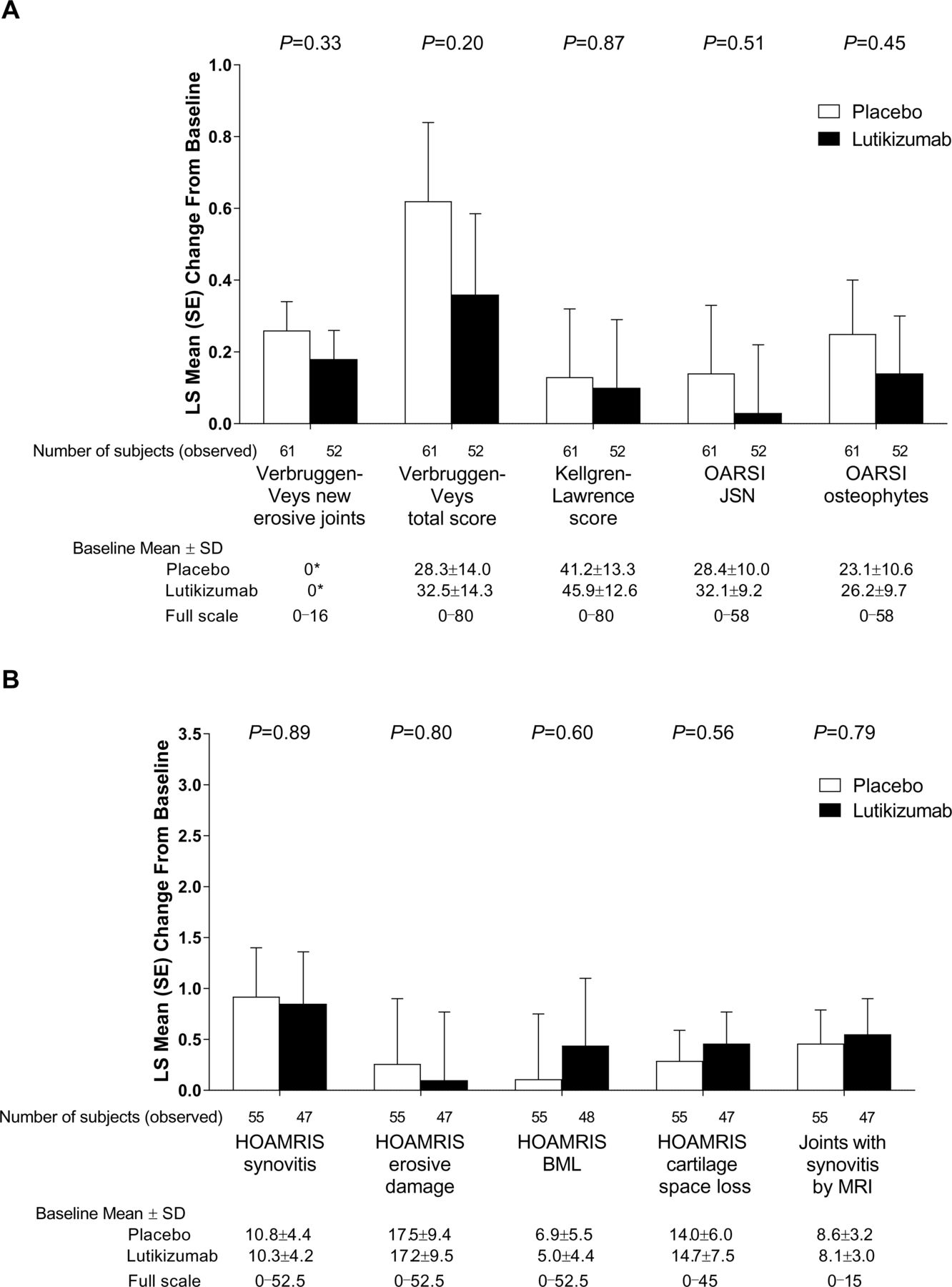
Changes from baseline in radiographic evidence of erosive joints (new E, E/R or R phase) and total score, as defined by Verbruggen et al and Verbruggen and Veys et al,33 35 were similar with lutikizumab and placebo at 26 weeks (figure 2A; table 2). Similar results were observed when using the Kellgren-Lawrence37 and OARSI scoring systems (figure 2A; table 2). MRI changes from baseline using the OMERACT/HOAMRIS system were not statistically significantly different for lutikizumab compared with placebo (figure 2B; table 2).

Assessment of radiographic endpoints (A) and MRI endpoints using the Outcome Measures in Rheumatology Clinical Trials/Hand Osteoarthritis MRI Scoring system (HOAMRIS) (B). *Defined by Verbruggen et al 33 as joints that entered the erosive, erosive with remodelling or remodelling phase but were normal, stationary or only starting to lose joint space at baseline. P values for lutikizumab versus placebo are from an analysis of covariance model adjusted for age group and Kellgren-Lawrence score as factors, and including baseline value as a covariate. BML, bone marrow lesions; JSN, joint space narrowing; LS, least squares; OARSI, Osteoarthritis Research Society International.
LS mean change from baseline to week 26 in scores for secondary or exploratory efficacy endpoints
Rescue medication use
Rescue medication use was similar between the treatment groups (online supplementary results). In a post-hoc analysis, there was no significant impact of disease symptom flares due to discontinuing NSAID use during the screening (washout) period, prior to enrolment and randomisation, on the primary endpoint of change from baseline to 16 weeks in AUSCAN pain. However, among subjects treated with lutikizumab, the magnitude of change in the primary endpoint was numerically but not statistically greater among subjects who were not using NSAIDs at the time of screening compared with subjects using NSAIDs at the time of screening.
Supplemental material
Pharmacokinetics
The mean serum concentrations of lutikizumab collected at the week 6 visit or later remained constant, suggesting attainment of steady-state lutikizumab serum levels at approximately twofold higher than concentrations collected at the week 2 visit, which is consistent with the observed half-life and accumulation ratio in phase I studies.30 The number of subjects with antilutikizumab antibodies at one or more study visits was 4 of 67 (6%) with placebo and 9 of 64 (14%) with lutikizumab. The magnitude of antilutikizumab antibody response was low and did not appear to impact the pharmacokinetic behaviour of lutikizumab.
Target engagement and pharmacodynamics
Lutikizumab treatment compared with placebo was associated with significant reductions in serum IL-1α and IL-1β levels, although serum levels of both cytokines were low and below the limits of quantification for some subjects (online supplementary figure 3 and online supplementary table 5). Lutikizumab significantly decreased the levels of neutrophils, hsCRP and serum C1M compared with placebo (figure 3). Serum C3M, CRPM and CTX-I were also reduced, although less robustly, in subjects treated with lutikizumab compared with placebo (online supplementary table 6). Changes in serum hyaluronic acid, serum PIIANP and urine CTX-II (corrected for urine creatinine) were similar in the lutikizumab and placebo treatment groups.
Supplemental material
Supplemental material
Supplemental material

{kind=link}
{kind=link}
{kind=link}
Mean hsCRP levels (A), neutrophil counts (B) and C1M levels (C) over time. *P<0.05, †P<0.01, ‡P<0.001, for lutikizumab versus placebo, one-way analysis of variance. C1M, metalloproteinase-degraded type I collagen; hsCRP, high-sensitivity C reactive protein.
Safety
Similar proportions of subjects receiving placebo or lutikizumab experienced an AE or a serious AE during the study (table 3). There were no serious infections during the study. Significantly more subjects in the lutikizumab treatment group had injection site reactions and neutropaenia (which was not associated with an increased rate of infection) compared with the placebo group.
Two subjects who received lutikizumab discontinued study treatment because of neutropaenia. One woman (aged 65 years) had a baseline ANC of 3.07×109/L, which decreased to 0.89×109/L after 15 days of treatment, at which time she discontinued lutikizumab. Another woman (aged 56 years) had a baseline ANC of 2.18×109/L, which decreased to 1.28×109/L after 29 days of treatment, at which time she discontinued lutikizumab.
There were significant mean reductions from baseline to final visit in neutrophil counts (figure 3B), platelet counts, white blood cell counts and alkaline phosphatase and increases in high-density lipoprotein cholesterol with lutikizumab but not placebo (online supplementary table 7). The mean low-density lipoprotein cholesterol (LDL-C) levels increased significantly from baseline with lutikizumab compared with placebo. Except for neutrophil counts and a single triglyceride level that was grade 3, the laboratory changes were all grade 1. Grade 2 and 3 laboratory neutropaenia events occurred in 14% and 5% of subjects, respectively, in the lutikizumab group and no subjects in the placebo group; there were no grade 4 neutropaenia events (table 3).
Supplemental material
Safety results
Discussion
In the present study, treatment with lutikizumab 200 mg was not significantly different from placebo for any efficacy endpoints. Lutikizumab had no effect on pain and function. Subjects treated with lutikizumab compared with placebo had similar radiographic progression by several measures, and the small differences were likely not clinically significant. These results suggest that targeting IL-1 may be ineffective in erosive HOA, in agreement with clinical studies in knee OA22 23 44 and a recent mouse study.45
It is unlikely that the lack of a lutikizumab treatment effect was due to insufficient levels of inflammation, because study subjects had moderate to severe levels of inflammation confirmed by tender and/or swollen interphalangeal joints and the presence of synovitis on MRI. Furthermore, the absence of a significant treatment effect could not be attributed to a lack of drug exposure, as lutikizumab serum levels were stable over time, and antidrug antibodies had no notable impact on the pharmacokinetics of lutikizumab, although drug levels in the local environment of the interphalangeal joints were not assessed. Additionally, lutikizumab markedly affected IL-1 levels and pharmacodynamic measures associated with inhibition of IL-1 (neutrophil counts and hsCRP levels). Furthermore, reductions in neutrophil counts and hsCRP were similar to those seen in a study of an IL-1R1 blocking antibody (AMG 108).23 Finally, data from a dose-range finding study of lutikizumab 25, 100 and 200 mg in subjects with knee OA strongly suggested that a dose of 200 mg every 2 weeks produced serum levels of lutikizumab that had maximal pharmacodynamic effects.44
Discontinuations due to AEs, neutropaenia and injection site reactions were more common with lutikizumab compared with placebo. AEs partly explain the higher overall discontinuation rate with lutikizumab; however, other unknown factors may have had a role. Reductions in neutrophil counts were not associated with increased rates of infection. These results are consistent with studies of other IL-1 inhibitors.23 46 47 LDL-C increased modestly on average with lutikizumab, as noted with other IL-1 inhibitors.48 49
Studies with other anti-inflammatory therapies have shown mixed results in erosive HOA. At least six studies have reported on the efficacy of anti-TNF agents in erosive HOA. In a 1-year, randomised, double-blind study (N=90), subcutaneous etanercept was not superior to placebo on a visual analogue pain scale at 24 weeks.11 However, in patients with more pronounced inflammatory symptoms who completed the study, etanercept was superior compared with placebo on pain and radiographically assessed structural damage.11 In a 1-year, randomised, double-blind study (N=60), no difference in erosive progression was seen after 12 months between groups who received adalimumab 40 mg subcutaneously every 2 weeks or placebo. However, in a post-hoc analysis, adalimumab was associated with significantly less erosive progression than placebo in patients with joints with soft-tissue swelling.10 In two other randomised, double-blind studies, adalimumab did not improve pain and other outcomes compared with placebo.50 51 In a single-blind study, 10 patients received monthly injections of infliximab or saline into affected joints (opposite hands in each patient). After 12 months of treatment, pain was significantly reduced by infliximab; radiographic lesion progression was reduced but not statistically different compared with saline.52 A 12-week, open-label study (N=12) with adalimumab suggested modest clinical efficacy.53
The strengths of the current study include its stringent enrolment of only patients with moderate to severe inflammation and the measurement of efficacy, multiple imaging scoring systems, and pharmacodynamics in the same subjects. However, several inherent limitations may have affected the findings. More than 26 weeks may have been needed to observe a structural effect of lutikizumab in patients with erosive HOA and to detect related pain and function improvement. Although pharmacodynamic measures such as the ANC suggest that IL-1 levels were maximally suppressed in the circulation by 200 mg of lutikizumab every 2 weeks,44 we did not measure hand joint synovial fluid levels of lutikizumab, IL-1α and/or IL-1β; therefore, IL-1α and IL-1β concentrations may not have been reduced locally in the joint to a level that improved pain and function or showed radiographic evidence of structural improvement. In addition, we have no adequate explanation for the low use of NSAIDs or analgesics, given the high average level of pain in patients at baseline.
In conclusion, despite adequate systemic lutikizumab pharmacodynamic effects, lutikizumab did not significantly improve clinical outcomes or imaging outcomes in patients with erosive HOA, compared with placebo, suggesting that targeting IL-1 may be ineffective for the treatment of erosive HOA.
Acknowledgments
We thank Gust Verbruggen of Ghent University Hospital for adjudicating discrepancies between readers in radiographic change scores. We thank Yan Chen of Spire Sciences for reading the MRIs. Medical writing support was provided by Richard M Edwards, PhD, and Michael J Theisen, PhD, of Complete Publication Solutions (North Wales, Pennsylvania, USA), a CHC Group company. Lutikizumab is an investigational product. AbbVie and the authors thank the patients who participated in the clinical trial and all study investigators for their contributions.
References
Footnotes
Handling editor Josef S Smolen
Presented at The results of this study were presented in part at the OARSI World Congress on Osteoarthritis (27–30 April 2017; Las Vegas, Nevada), the European League Against Rheumatism Congress (14–17 June 2017; Madrid, Spain) and the American College of Rheumatology Annual Meeting (4–8 November 2017; San Diego, California).
Contributors All authors contributed to the development of the content. All authors and AbbVie reviewed and approved the final manuscript, and the authors maintained control over the final content.
Funding AbbVie funded this study (NCT02384538). AbbVie funded the medical writing support.
Competing interests AbbVie contributed to the design of the study and was involved in the collection, analysis and interpretation of the data, and in the writing, review and approval of the publication. MK has received grant/research support from Pfizer and been a consultant for AbbVie, GlaxoSmithKline, Merck and Levicept. CP is an employee of Spire Sciences, and is on the speakers' bureau for Amgen and Bristol-Myers Squibb. IKH has been a consultant for AbbVie. FK has no conflicts of interest to declare. RMF has received grant/research support from and has been a consultant for AbbVie. FB has been a consultant for AbbVie, Pfizer and Regeneron. DvdH has been a consultant for AbbVie, Amgen, Astellas, AstraZeneca, Bristol-Myers Squibb, Boehringer-Ingelheim, Celgene, Daiichi, Eli Lilly, Galapagos, Gilead, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi and UCB; she is Director of Imaging Rheumatology. PB is a former employee of PAREXEL, which performed work for this study under contract to AbbVie. RW has been a consultant for AbbVie. SC, LW, WL, GL, YF, MS, JKM and MCL are employees of AbbVie and may own AbbVie stock and/or stock options. SF is a former employee of AbbVie and may own AbbVie stock and/or stock options.
Patient consent Not required.
Ethics approval The protocol was approved by institutional review boards at each study site.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymised, individual and trial-level data (analysis data sets), as well as other information (eg, protocols and clinical study reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. These clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
Author note PB was affiliated to Scientific and Medical Services, PAREXEL, and SF was affiliated to Exploratory Statistics, Data Science and Statistics, AbbVie, at the time of this study.