Article Text

Abstract
Objective Salmonella enterica infections can lead to Reactive Arthritis (ReA), which can exhibit an association with human leucocyte antigen (HLA)-B*27:05, a molecule prone to misfolding and initiation of the unfolded protein response (UPR). This study examined how HLA-B*27:05 expression and the UPR affect the Salmonella life-cycle within epithelial cells.
Methods Isogenic epithelial cell lines expressing two copies of either HLA-B*27:05 and a control HLA-B*35:01 heavy chain (HC) were generated to determine the effect on the Salmonella infection life-cycle. A cell line expressing HLA-B*27:05.HC physically linked to the light chain beta-2-microglobulin and a specific peptide (referred to as a single chain trimer, SCT) was also generated to determine the effects of HLA-B27 folding status on S. enterica life-cycle. XBP-1 venus and AMP dependent Transcription Factor (ATF6)-FLAG reporters were used to monitor UPR activation in infected cells. Triacin C was used to inhibit de novo lipid synthesis during UPR, and confocal imaging of ER tracker stained membrane allowed quantification of glibenclamide-associated membrane.
Results S. enterica demonstrated enhanced replication with an altered cellular localisation in the presence of HLA-B*27:05.HC but not in the presence of HLA-B*27:05.SCT or HLA-B*35:01. HLA-B*27:05.HC altered the threshold for UPR induction. Salmonella activated the UPR and required XBP-1 for replication, which was associated with endoreticular membrane expansion and lipid metabolism.
Conclusions HLA-B27 misfolding and a UPR cellular environment are associated with enhanced Salmonella replication, while Salmonella itself can activate XBP-1 and ATF6. These data provide a potential mechanism linking the life-cycle of Salmonella with the physicochemical properties of HLA-B27 and cellular events that may contribute to ReA pathogenesis. Our observations suggest that the UPR pathway maybe targeted for future therapeutic intervention.
- ankylosing spondylitis
- infections
- reactive arthritis
- spondyloarthritis
- lipids
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Misfolding of the human leucocyte antigen B27 allele (HLA-B27), known to induce the unfolded protein response (UPR),1 2 has been proposed to explain why HLA-B27 drives the inflammatory arthritic disorder ankylosing spondylitis (AS). Protein misfolding within the ER can disrupt ER homeostasis and trigger the UPR as measured by activation of the XBP-12 and ATF6 pathways,1 and in patients with AS by upregulating the UPR effector gene, HRD1.3
The clinical manifestation of Reactive Arthritis (ReA) after Salmonella enterica infection has a mixed association with HLA-B27.4 Some studies suggest that HLA-B27-positive individuals exhibit increased susceptibility to ReA5–7or increased risk of Salmonella infection,8 while others have found no strong association.9–11 S. enterica grows within a specialised membrane-bound compartment termed the Salmonella-containing vacuole (SCV) prior to cellular escape and dissemination.12–14 The mechanistic link between intracellular bacteria and HLA-B27 is unknown, but mammalian cell lines expressing HLA-B27 can harbour higher numbers of internal Salmonella.15–17
We have previously shown that fusing the HLA-B27 heavy chain (HC), beta-2-microglobulin (β2m) and presented peptide together within a single chain trimer (SCT) significantly reduces misfolding relative to the original HC.18 Here we have used the SCT to test the hypothesis that HLA-B27 misfolding impacts on endocellular growth and localisation of S. enterica in infected cells. We also tested the extent to which S. enterica influences activation of both the XBP-1-and ATF6-mediated UPR pathway.
Materials and methods
UPR induction
UPR responses were induced with tunicamycin (TUN), thapsigargin (TPG), MG-132 or calcimycin (A23187) from Calbiochem, with appropriate vehicle (dimethyl sulfoxide (DMSO) alone) controls.
Transfection of UPR reporter constructs
Polyethylenimine (JetPrime) was used to transfect cells with the UPR reporter plasmids ΔDBDXBP-1 venus (v) and ATF6-FLAG19 20 following the manufacturer’s conditions. Cells were fixed at the desired postinfection (pi) time points for 10 min with 3.8% paraformaldehyde (PFA: pH 7.4) and fluorescence was measured using LSR2 and LSR Fortessa flow cytometers (BD Biosciences), and the data were analysed using FlowJo V.8.7.3 software.
S. enterica cfu enumeration and microscopy
Colony-forming unit (cfu) enumeration was performed by lysing cells in 1% Triton X-100/phosphate buffered saline (PBS). Lysates were serially diluted into 1% bovine serum albumin/0.1% Tween-80% and plated on Luria Broth (LB) agar at room temperature for ~16 hours. Each experimental condition was performed in triplicate and each plating in duplicate. For microscopic analysis, coverslips containing infected cells were washed with 1× PBS, fixed for 10 min with 3.8% PFA (pH 7.4), washed twice with 1× PBS and stored at 4°C.
UPR-mediated membrane expansion during infection
Glibenclamide BODIPY FL (green; Invitrogen) was used to quantitate endoreticular membrane size and localisation. Henrietta Lacks (HeLa) cells were treated with UPR-inducing drugs and labelled with glibenclamide according to the manufacturer’s protocol. Labelled cells were analysed by fluorescence activated cell sorting (FACS). Cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) and visualised by fluorescence microscopy. For control and drug-treated cells, equivalent exposures were collected.
To determine endoreticular-derived membrane expansion during infection, HeLa cells were grown either on sterile glass, infected with S. enterica Typhimurium expressing mCherry (see online supplementary materials and methods) and stained with glibenclamide green. Cells were fixed, washed and counterstained with DAPI, followed by fluorescence microscopy or automated confocal analysis. Images were acquired by an Opera LX (PerkinElmer) plate reader with a confocal microscope (NA=0.6, 40× air objective). Exposure times were 100 ms for the DAPI channel (365 nm), 2000 ms for the ER channel (488 nm) and 2000 ms for the Salmonella channel (561 nm). Camera pixels were binned by two resulting in a pixel size of 0.323×0.323 µm, and 4800 images were acquired per 96-well plate (50 images per well), which were processed in one batch using the same image analysis pipeline, algorithms and parameters (see online supplementary materials and methods for analysis of glibenclamide mean fluorescence intensity (MFI)).
Supplemental material
Results
XBP-1 and ATF6 activation following Salmonella infection
We used our previously described epithelial cells with identical sites of transgene integration (and therefore isogenic) expressing physiological levels of HLA-B273 (online supplementary figure S1A–E). Control cell lines encoding HLA-B*35:01 HC (HLA-B35.HC) transgene or the FRT vector alone (referred to as Empty (E)84) were generated (integration at the same two loci). HLA-B27 and HLA-B35.HC lines were incubated with or without the influenza nucleoprotein peptide 383–391 (NP383-391). The ability of HLA-B27 to present cognate peptide to NP383-391-specific CD8+ cytotoxic T lymphocytes (CTL) was confirmed by an increase in CD107a cell surface expression (online supplementary figure S1F, top right panel). Two-dimensional immunoblotting of cell lysates revealed HLA-B27.HC can form HC-dimers under these experimental conditions (figure 1A, arrow top panel), but only monomeric HLA-B35.HC was detected (figure 1A, bottom panel).
Supplemental material

Salmonella exhibits enhanced replication in the presence of HLA-B27 and can activate XBP-1 and ATF6. (A) Two-dimensional isoelectric focusing of lysates from B27.HC cells and immunoblotted with anti-V5 pK demonstrates dimer formation (arrow) (top panel), while B35.HC does not form dimers. (B) HLA.B27.HC-expressing cells, transfected with the XBP-1ΔDBDv reporter construct (UPR leads to a 26nt intronic sequence excision, leading to the expression of the venus fluorescent protein) and treated with increasing concentrations of TPG exhibit a reduced UPR threshold. (C) The fold increase in ΔDBDXBP-1v activation following infection of HeLa cells with ST.mCherry at MOI 3:1, 50:1 and 100:1. (D) The fold increase in ΔDBDXBP-1v activation following infection of HeLa cells with ST.mCherry. Non-infected cells (mCherry-negative) from the same well expressing ΔDBDXBP-1v were used as non-infected controls to calculate the fold increase in XBP-1 activation in the infected cells (mCherry-positive). No induction of XBP-1v activation was seen in cells treated with heat-killed Salmonella enterica Typhimurium (HTST), Lipopolysaccharide (LPS) and flagellin (FGN). (E) Activation of endogenous XBP-1s was demonstrated by qPCR. All cycle threshold values were normalised to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and data plotted as a fold induction of XBP-1s mRNA compared with levels detected at baseline. (F) The fold increase in ATF6 activation as measured by the cleaved cytosolic-FLAG tagged domain, following infection of HeLa cells with ST.mCherry. (G) Immunoblot with anti-FLAG tag antibody of AMP dependent transcription factor 6 (ATF6) following infection in two separate experiments (INF1 and INF2) compared with non-infected (NI) cells. Inactivated (i) and activated (a) ATF6 are denoted by arrows. For FACS analysis of ΔDBDXBP-1v activation (n=3) and densitometry analysis ofATF6 activation (n=3) in infected cells mean values ±SEM are shown and ANOVA was performed (p <0.0001 for both figures) with Tukey’smultiple comparison post-test. Significant differences between groups areindicated P< 0.05 (*), P< 0.01 (**) and P< 0.001 (***) (H) Relative fold increase of bacteria recovered from four individual experiments. Fold increases in Salmonella recovered were calculated based on cfu recovered from control E84. There in an average 4.2-fold increase in bacteria recovered from Salmonella-infected HeLa.B27.HC cells relative to the E84 and HLA-B35.HC control lines 24 hours post infection. The Mann-Whitney test was used to compare cfu recoveries between HLA-B35-expressing and HLA-B27-expressing cell lines and the exact p values were calculated (*p=0.0286). ATF6, AMP dependent transcription factor 6;cfu, colony-forming unit; E84, empty 84; HC, heavy chain; HeLa, Henrietta Lacks; HLA, human leucocyte antigen; LPS, Lipopolysaccharide; MOI, multiplicity of infections; TPG, thapsigargin; UPR, unfolded protein response; v,venus.
HLA-B27 expression can activate XBP-12 21; therefore, we determined whether our cell lines had altered sensitivity to UPR induction. Using the XBP-1vΔDBD reporter, which on splicing the 26 bp intronic sequences leads to GFP expression,19 we observed no induction of the UPR within these cell lines under normal culture growth conditions. Reasoning the expression of HLA-B27 could alter the threshold at which the UPR could be activated, we monitored XBP-1vΔDBD activation with increasing concentrations of the UPR-inducing TPG agent. HLA-B27-positive cells had an altered threshold for UPR induction, while the E84 and B35 lines did not exhibit significant differences (figure 1B).
Next, we determined whether Salmonella affected the UPR in the absence of HLA-B27 expression. Cells were infected with S. enterica expressing mCherry (referred to as ST.mCherry), and activation of ΔDBDXBP-1v was monitored following various multiplicity of infections, including 3:1, 50:1 and 100:1 over 1, 4, 8 and 24 hours. XBP-1s activation was determined by the fold increase in MFI above the uninfected ΔDBDXBP-1v transfected cells. Infection led to enhanced XBP-1s protein levels 8–24 hours pi (figure 1C). Toll like receptors (TLR) ligands LPS and flagellin did not activate the ΔDBDXBP-1v reporter and heat-killed bacteria had minimal impact (figure 1D). Endogenous XBP-1s mRNA levels were then quantified and quantitative (q)PCR of XBP-1s transcript levels showed increased expression 8–16 hours pi, demonstrating activation of the IRE-1 pathway (figure 1E). We also examined the impact of infection on the ATF6 pathway. Cells were transfected with ATF6-FLAG, and the cleaved cytosolic-FLAG tagged domain was quantified by immunoblotting pi. The peak cleavage of the ATF6 cytosolic domain was observed 12 hours pi (figure 1F–G) with a 1.5-fold to 2.5-fold increase in activation in infected compared with non-infected cells. HeLa.B27.HC-expressing and HeLa.B35.HC-expressing cell lines were then tested for their ability to support bacterial growth. Cells were infected with S. enterica for 24 hours prior to enumeration of cfu. A fourfold to fivefold increase in cfu from HLA-B27.HC-expressing cells was observed compared with cells expressing HLA-B35.HC or E84 controls (data not shown) (figure 1H).
XBP-1 pathway is required for efficient S. enterica Typhimurium replication
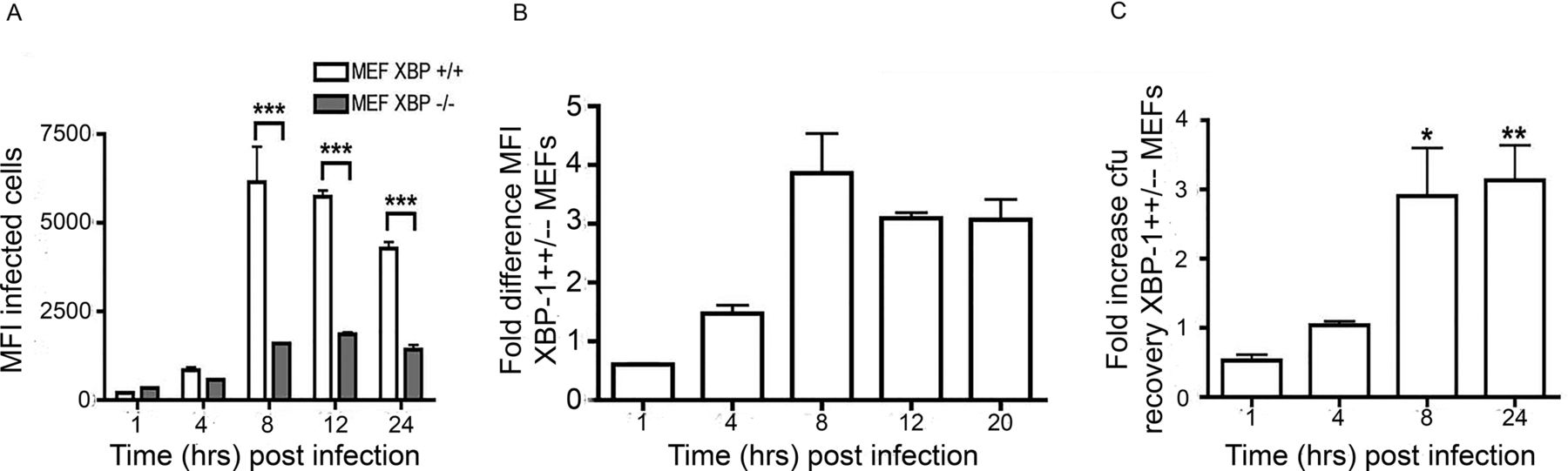
Our observations suggested that activation of the XBP-1 pathway plays a significant role in S. enterica growth and replication. To support our hypothesis, we analysed S. enterica replication in XBP-1-deficient mouse embryonic fibroblasts (MEF). XBP-1+/+ and XBP-1−/− MEF cell lines were infected with ST.mCherry, harvested and analysed by flow cytometry. Importantly the MFI, which is a measure of the average number of bacteria per cell, for both cell types were similar at 1 and 4 hours pi, indicating no significant differences in either bacterial invasion or early infection stages. However, when the bacteria began replication, the XBP-1−/− MEF cell line exhibited significantly lower MFI values than the XBP-1+/+ cell line, indicating reduced bacterial growth (figure 2A). XBP-1+/+ cells had approximately threefold more bacteria than in XBP-1−/− cells during the replication phase (8–24 hours pi) (figure 2B–C).

Salmonella requires an intact XBP-1 pahway for efficient replication. (A) Graph comparing the MFI of infected cells. The MFIs of both cells types were similar at early time points (1 and 4 hours pi). When the bacteria enter the log phase replication, the XBP-1−/− cells show much lower MFI values than XBP-1+/+ cells. The mean mCherry MFI values±SEM are shown (n=3) and analysis of variance was performed (p<0.0001) with Tukey’s multiple comparison post-test to determine if there were significant differences between individual groups (***p<0.001). (B) The fold difference in the MFI of the XBP-1+/+/XBP-1−/− cells at the various time points pi. (C) Recovery of viable bacteria from infected cells shows similar differences between the XBP-1+/+ and XBP-1−/− cells at 8 and 24 hours pi. The mean fold differences in cfu values (n=5 or 6)±SEM are shown. The MannWhitney test was used to compare cfu recoveries.Recoveries between XBP-1+/+ andXBP-1-/- cells and exact p values calculated (* P = 0.015 and ** P=0.031). cfu, colony-forming unit; MEFs, mouse embryonic fibroblasts; MFI, mean fluorescence intensity; pi, postinfection.
Enhanced bacterial replication is linked to UPR-induced lipid metabolism
Next we wished to test the hypothesis that UPR promotes Salmonella replication. To mimic UPR conditions, HeLa cells were pretreated with the following UPR-inducing pharmacological agents: A23187 (0.5 mM), MG132 (0.5 µg/mL), TUN (0.5 µg/mL) and TPG (200 nM) (see figure 3A for mode of action). HeLa cells were treated with DMSO (vehicle control) or the respective UPR inducer 16 hours prior to infection with ST.mCherry, and the fold increase in infected cells over the control was determined 24 hours pi. Overall the data indicate that a UPR environment leads to an increase in intracellular Salmonella (figure 3B).

Salmonella exhibits enhanced recovery from cells undergoing UPR. (A) Pharmacological UPR-inducing agents and their mode of action. A23187 is a Ca2+ ionophore that disrupts intracellular Ca2+ levels. Thapsigargin is a non-competitive inhibitor of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump leading to reduced Ca2+ endoplasmic reticulum (ER) concentrations. Tunicamycin inhibits the addition of carbohydrate (CHO) moities to newly synthesised proteins. MG132 inhibits proteasome-mediated degradation leading to accumulation of misfolded proteins within the ER. Triacin C is also shown and inhibits de novo lipid synthesis. (B) Treatment of cells with UPR-inducing drugs increases the number of intracellular bacteria 24 hours pi. Cells were treated with A23187 (0.5 mM), MG132 (0.5 µg/mL), TPG (200 nM) and TUN (0.5 µg/mL). The mean fold increases in mCherry MFI values±SEM are shown (n=3). ANOVA was performed on mCherry MFI values (p<0.0001) with Tukey’s multiple comparison post-test to determine significant differences between individual groups (*p<0.05m, ***p<0.001. (C–D) TPG-treated cells exhibit significant increases in MFI values of the infected cells when compared with dimethyl sulfoxide (DMSO) controls (C) and in the fold difference in MFI values between the DMSO-treated and TPG-treated cells (D) in a concentration-dependent manner. The mean MFI values±SEM (n=3) are shown and ANOVA was performed (p<0.0001) with Tukey’s multiple comparison post-test used to determine if there were significant differences between DMSO and all drug-treated groups at 24 hours pi (***p<0.001). (E) Recovery of viable bacteria from infected cells exhibits similar fold differences between the DMSO-treated and TPG-treated cells infected at 24 hours pi. The mean fold differences in cfu values (n=6)±SEM are shown. The Mann-Whitney test was used to compare cfu recoveries between DMSO-treated and TPG-treated cells and the exact p values were calculated (*p=0.004, **p=0.015). ANOVA, analysis of variance; cfu, colony-forming unit; ER, endoplasmic reticulum; MFI, mean fluorescence intensity; pi, postinfection; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; TPG, thapsigargin; TUN, tunicamycin; UPR, unfolded protein response.
TPG appeared to have a more pronounced effect on Salmonella replication and also has the most direct effect on ER homeostasis (figure 3A). We employed TPG further to determine the effect of a pre-existing endoplasmic reticulum (ER) stress environment. HeLa cells were treated with TPG at 25, 100 and 400 nM 16 hours prior to infection. TPG pretreated cells showed significant increases in the MFI of infected cells when compared with the controls (figure 3C). A fourfold to sevenfold increase in TPG-treated cells was recorded 24 hours pi (figure 3D). Interestingly, flow cytometry revealed no significant difference in the percentage of HeLa cells infected with increasing TPG concentrations (online supplementary figure 2). Direct quantification of bacterial replication by cfu recovery also demonstrated significant increase in bacterial numbers in TPG-treated cells at 8 and 24 hours pi (figure 3E). Depending on the signals and duration, UPR can lead to production of proapoptotic or antiapoptotic factors. The observed increase in bacterial counts 8 hours pi may be due to the production of antiapoptotic factors. To assess the level of cell death, activation of caspases was followed by staining with the fluorescein isothiocyanate (FITC) conjugated pan caspase detection reagent FLICA and the live dead dye 647. Treatment of cells with UPR-inducing drugs did not alter activation of caspases or induction of cell death during infection, indicating that the observed enhanced bacterial counts post-UPR are not linked to increased cellular death (online supplementary figure 3).
Supplemental material
Supplemental material
Salmonella replication and intracellular niche development require a source of membrane, which would depend in part on lipid biosynthetic pathways, which can be activated and/or enhanced by the UPR. We therefore inhibited de novo synthesis of long-chain fatty acid synthesis during the induction of the UPR with Triacin C (TRC), a potent inhibitor of long fatty acyl CoA synthetase isoforms 1, 3 and 4. HeLa cells were treated with DMSO, TRC (200 nM) and TPG (200 nM) or cotreated with TRC (200 nM) and TPG (200 nM) 16 hours prior to infection. TRC treatment prior to infection did not significantly alter the percentage of cells infected either in the presence or absence of TPG (online supplementary figure 2). Cells treated with TPG alone showed significant increases in MFI values of the infected cells when compared with the DMSO or TRC controls (figure 4A). However, cotreatment of cells with TRC and TPG significantly reduced the effect of the UPR induction on the levels of intracellular bacteria 24 hours pi (figure 4A,B). These observations were confirmed by recovery of viable bacteria from similarly infected HeLa cells (figure 4C), indicating inhibition of lipid metabolism during UPR induction reduces the increase in intracellular bacteria observed at late time points.
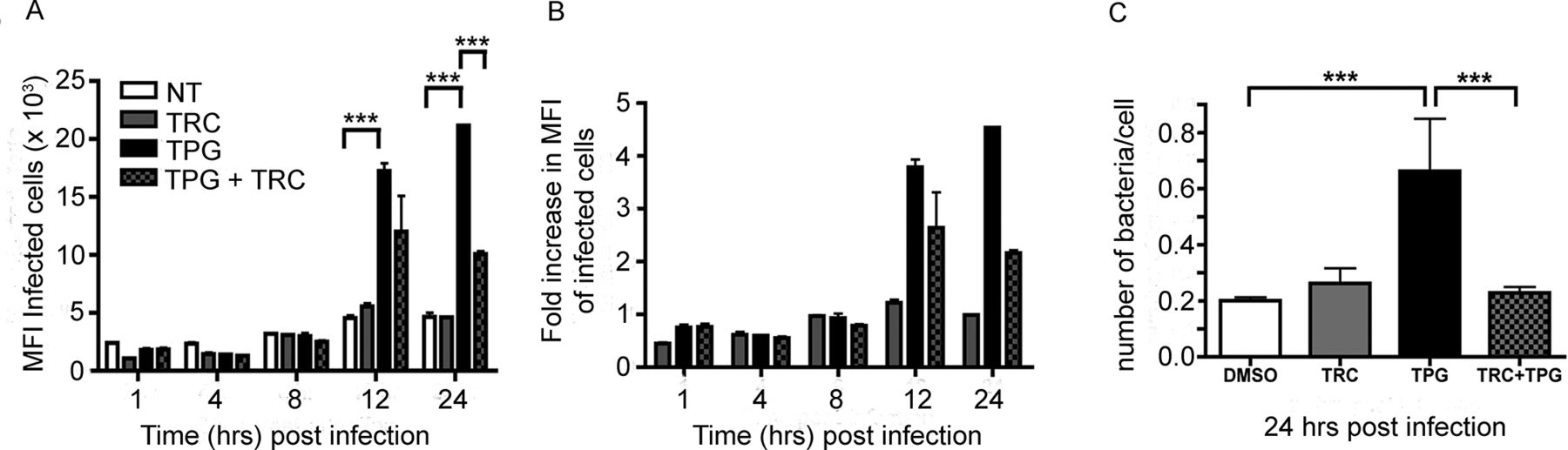
Salmonella requires de novo lipid synthesis during enhanced recovery from cells undergoing UPR. (A) HeLa cells were treated with DMSO, TRC (200 nM) and TPG (200 nM) or cotreated with 200 nM TRC and 200 nM TPG at 16 hours prior to infection with ST.mCherry. TPG-treated cells exhibit significant increases in the MFI values of the infected cells when compared with DMSO or TRC controls. Cotreatment of cells with TRC and TPG significantly reduces the effect of the TPG on the levels of intracellular bacteria at 24 hours pi. The mean mCherry MFI values±SEM are shown (n=3) and analysis of variance was performed (p<0.0001) with Tukey’s multiple comparison post-test to determine significant differences between individual drug treatment groups (***p<0.001). (B) The fold difference in the MFI values of the DMSO, TRC, TPG and TRC/TPG treated cells. (C) Observation by FACS was confirmed by recovery of viable bacteria from similarly treated and infected cells. Colonies were counted and the number of bacteria present per cell in each sample was calculated (n=12). The Mann-Whitney test was used to compare cfu recoveries between DMSO and TPG or TPG and TRC/TPG cotreated cells and the exact p values were calculated (***p=0.0001). cfu, colony-forming unit; DMSO, dimethyl sulfoxide; HeLa, Henrietta Lacks; MFI, mean fluorescence intensity; NT, not treated; pi, postinfection; TPG, thapsigargin; TRC, Triacin C; UPR, unfolded protein response.
Salmonella infection leads to endoreticular membrane expansion. To determine whether the increase in intracellular bacteria from cells undergoing the UPR was dependent on replication within the SCV, we used the isogenic S. enterica Typhimurium ΔsifA mutant, which can infect but is impaired in intracellular growth and can escape the SCV.22–24 HeLa cells were treated with DMSO or 200 nM TPG 16 hours prior to infection with ST.mCherry or the isogenic ΔsifA mutant. ST.mCherry exhibited significant increases after TPG treatment as already described. In marked contrast, UPR-induced cells infected with the ΔsifA mutant demonstrated no increase in bacterial numbers (figure 5A). Thus, intracellular localisation within the SCV is required for UPR-mediated effects on bacterial replication.
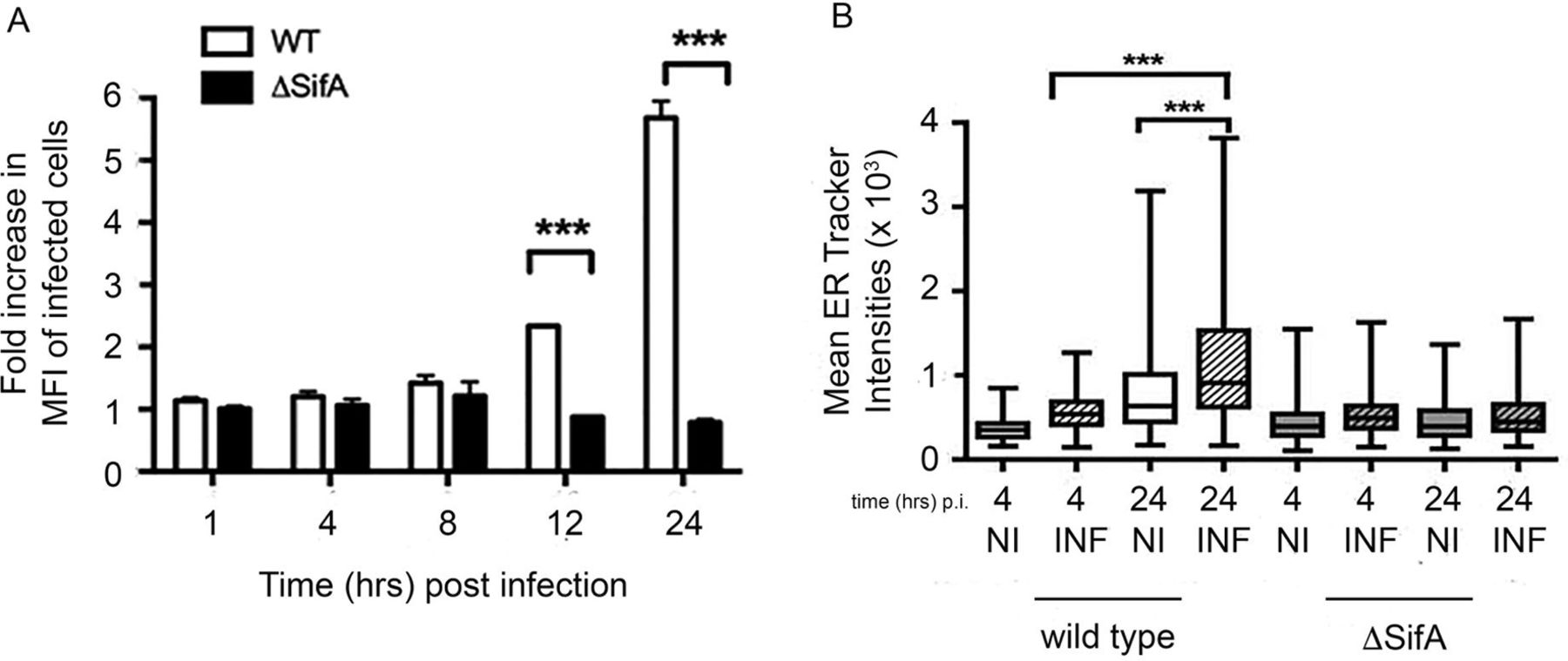
Salmonella infection can increase endoreticular membranes and exhibit altered cellular location in the presence of HLA-B27. (A) Increase in the number of intracellular bacteria in cells treated with UPR-inducing drugs is dependent on the intracellular replication of bacteria within the SCV. HeLa cells were treated with DMSO or 200 nM TPG at 16 hours prior to infection with Salmonella enterica Typhimurium 12 023 or ΔsifA strains expressing mCherry. Wild-type (WT) bacteria, which replicate within the SCV, show increases in intracellular bacteria in the TPG-treated samples, while those infected with the ΔsifA mutant show no increase in the TPG-treated cells. (B) HeLa cells were infected with WT or ΔsifA ST.mCherry, stained with ER tracker (green) at 4 and 24 hours pi and analysed by confocal microscopy using an Opera LX plate reader. Quantification of endoreticular membrane content in infected cells. MFI of glibenclamide staining in infected (INF) and non-infected (NI) cells were compared at 4 and 24 hours pi with either WT or ΔsifA 12023. MFI values±SEM are shown (n=135–1476). For (B) statistical analysis was performed using the Kruskal-Wallis test (p<0.0001) and was performed with Dunn’s multiple comparison post-test to determine significant differences between individual groups (***p<0.001). DMSO, dimethyl sulfoxide; ER, endoplasmic reticulum; HeLa, Henrietta Lacks; HLA, human leucocyte antigen; MFI, mean fluorescence intensity; pi, postinfection; SCV, Salmonella-containing vacuole; TPG, thapsigargin; UPR, unfolded protein response.
As UPR activation is associated with endoreticular membrane expansion and lipid metabolism,25 26 we determined whether Salmonella infection could lead to expansion of endoreticular membrane during the replication/growth phase of its life-cycle using glibenclamide green ER tracker dye as a marker for endoreticular membranes. HeLa cells were infected with either wild-type or ΔsifA ST.mCherry as a control strain. At 4 and 24 hours pi, cells were stained with glibenclamide green ER tracker dye and analysed by automated confocal microscopy. The levels of glibenclamide-labelled membrane in infected cells were quantified and compared with uninfected cells. An increase in glibenclamide labelling in cells infected with wild-type but not sifA-deficient bacteria was observed (figure 5B), which supports our observation that XBP-1 is not activated by ΔsifA bacteria (data not shown).
The folding status and expression of HLA-B27 alter Salmonella replication and cellular localisation
To address the role of the folding status of HLA-B27, we generated an HLA-B27 molecule fused to the light chain β2m and an HLA-B27-specific peptide derived from the influenza nucleoprotein (referred to as HLA-B27.SCT). As a control we also generated a similar fusion protein for HLA-B35 with EBNA1 peptide (figure 6A). The HLA-B27 and HLA-B35.SCTs were transfected into the original FLIPIN HeLa founder line. Functional activity of the HLA-B27.SCT was determined by incubation±NP383-391 peptide. The HLA-B27.SCT line was an effective CTL target in the absence (figure 6B, top left panel) and presence (figure 6B, top right panel) of exogenously added peptide, whereas the HLA-B35.SCT line did not activate HLA-B27-NP-restricted CTL lines (figure 6B, bottom left and right panels). We next determined whether HLA-B27.SCT could form dimeric conformations. Both cell lines were treated with N-ethylmaleimide (NEM) and lysates were seperated by charge and Mw as described previously. Immunoblotting with the anti-V5 tag antibody pK revealed that neither the HLA-B27 SCT or HLA-B35 SCT lines form high Mw conformers (figure 6C, top and bottom panels, respectively).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Salmonella exhibits altered cellular location in the presence of folding and misfolding HLA-B27. (A) Schematic of the MHC class I SCT used in this analysis. (B) HLA-B27.SCT (top left and right panels) can activate NP-specific T cell clones and act as efficient targets in the absence of exogenous NP peptide (top left panel). HLA-B35.SCT does not activate NP-B27-specific CTLs (bottom left and right panels). (C) Two-dimensional isoelectric focusing of lysates from B27.SCT and B35.SCT cells and immunoblotted with anti-V5 pK demonstrate no dimer formation. (D) Expression of HLA-B27 in the context of the SCT molecule reverses the enhanced bacterial recovery phenotype observed in HLA-B27 HC-expressing cells. ANOVA was performed (p=0.0033) with Tukey’s multiple comparison post-test to determine significance between individual groups (*,**p<0.05). (E) Differences in bacteria recovered at later time points is not due to increased adhesion or invasion of HeLa.B27.HC by ST.GFP. Flow cytometric analysis of cells infected with ST.GFP over time shows no observed difference in the number of cells infected or relative number of bacteria per cell (as determined by MFI) until later infection time points, that is, >8 hours pi. Data are presented relative to the results from control E84 cells. Two-way ANOVA was performed (p<0.0001) with multiple t-tests to determine significance between individual groups (****p<0.0001). (F) Salmonella (green) resides in SCVs associated with the Golgi apparatus, stained with giantin (red) in HeLa.FLP (E84, control i) and HeLa.B35.HC (ii). HeLa.B27.SCT-expressing cells (iv) exhibit no altered localisation of Salmonella when compared with HLA-B27.HC (iii). Arrow heads highlight Salmonella localisation. Nuclei are stained with DAPI (blue). ACTL, cytotoxic T lymphocytes; ANOVA, analysis of variance; cfu, colony-forming unit; APC, allophycocyanin; DAPI, 4′,6-diamidino-2-phenylindole; E84, Empty 84; FLP, flip; GFP, green fluorescent protein; HC, heavy chain; HLA, human leucocyte antigen; MFI, mean fluorescence intensity; MHC, major histocompatibility complex; NP, nucleoprotein peptide; PE, phycoerythrin; pi, postinfection; SCT, single chain trimer; SCV, Salmonella-containing vacuole.
Next, we wished to know whether HLA-B27.SCT cells could support enhanced Salmonella replication. HLA-B27.SCT, along with HLA-B35 and B27.HC lines, were infected with ST.Green fluorescent protein (GFP). CFU recovery, determined 24 hours pi, demonstrated enhanced numbers of bacteria in the B27.HC but not in the B27.SCT line (figure 6D).
As Salmonella survival can correlate with their intracellular localisation,27 we tracked ST.GFP within the different HLA-class I expressing cell lines using confocal microscopy. Following infection, cells were costained for the trans-Golgi specific marker giantin (red) and the nucleus with DAPI (blue) (figure 6E). In the E84 and HLA-B35.HC cell lines, we detected ST.GFP concentrated in juxtaposition to the Golgi apparatus, which reflects Salmonella within the SCV. Surprisingly, in the presence of HLA-B27.HC, we noted that Salmonella markedly do not reside in close proximity to the Golgi, but instead was located more within the periphery (figure 6F, panel ii). In contrast, in infected B27.SCT cells (figure 6F, panel iv), the bacteria resided in similar locations to the B35 and E84 cells (figure 6F, panel iii), suggesting that bacterial location is associated with enhanced replication, in a process influenced by the folding efficiency of HLA-B27.
Discussion
Why Salmonella exhibits an association with ReA and HLA-B27 remains undetermined. Here we have analysed Salmonella growth where HLA-B27 misfolding and the UPR are limiting parameters. Our study demonstrates that it is not the expression of HLA-B27 alone that results in enhanced bacterial replication, but HLA-B27 misfolding, which influences the ER stress environment. Our demonstration that HLA-B27 expression can reduce the threshold of ER stress induction and that Salmonella can induce the UPR provides key additional insight as to why such bacteria manipulate and exploit ER stress pathways to their benefit.
In the presence of misfolding HLA-B27, Salmonella predominantly resides in an altered peripheral cellular localisation. Thus HLA-B27 may alter SCV biogenesis and intracellular movement. Maturation of the SCV and bacterial cellular localisation can determine survival and replicative capability of Salmonella.14 22 27 28 During the early stages of infection, the SCV migrates by following an endosomal maturation route, to a juxtanuclear location associated with the microtubule organising centre and the Golgi apparatus in epithelial cells.13 14 It is possible that the bacteria fail to form an SCV or exit the SCV more rapidly in the presence of misfolding HLA-B27, or escape from the SCV and randomly redistribute throughout the cytoplasm. Alternatively, Salmonella could initially occupy SCVs in juxtaposition to the Golgi and the movement to the periphery is enhanced in the presence of HLA-B27.
HLA-B27.HC expression and endocellular S. enterica growth both independently cause activation of the XBP-1 ER-stress pathway. XBP-1 activation peaks at 8–16 hours pi (figure 1D,E), which correlates with enhanced bacterial numbers in cells undergoing ER stress and/or expressing HLA-B27.HC with a propensity to misfold. Activation of both XBP-1 and ATF6 coincides with the replication/growth phase of S. enterica Typhimurium. During this phase Salmonella membrane modifications have been reported to be at their peak,29 which might therefore lead to UPR activation. We do not currently know the complete cellular stress response to Salmonella in our model cell system, but plan to address this by RNA-Seq to map other significant differences in how HLA-B*27:05-expressing and HLA-B*35:01-expressing cells respond after infection. However, taken together, data reported here suggest the ER stress consequences of HLA-B27 misfolding provide a favourable environment for replication of S. enterica within the HeLa cells. Interestingly, Salmonella infections can affect interleukin-23 production,30 a cytokine that has been implicated to be important in the disease phenotype of spondyloarthropathies.31 It is possible that the co-occurrence of Salmonella and HLA-B27 could have a cumulative or multiplicative effect on the UPR, which could explain the enhanced risk of ReA and/or an increased risk of symptomatic Salmonella infection in HLA-B27-positive individuals in a population exposed to infection.5 6 8 However, the effects of HLA-B27 on Salmonella may depend on the temporal activation of the UPR.32 Our observations with pre-existing UPR activation do suggest that these aforementioned factors could indeed influence Salmonella replication.
The origin of the membrane that makes up the growing SCV remains poorly defined, but ER membrane markers have been reported in SCVs. The ER membrane-bound markers calnexin and protein disulfide isomerase (PDI) were demonstrated to codistribute with SCVs and up to 20% of intracellular bacteria.33 34 Proteome analysis of host cell membranes modified by Salmonella indicated that ER membranes can be redirected to their intracellular niche.29 UPR activation can regulate ER membrane by increasing phospholipids and ER protein levels, as well as modulating fatty acid, sphingolipid, phospholipid and sterol metabolism, which ultimately lead to expansion of ER membranes.25 26 26 35 36 Our experiments with TRC suggest that the long-chain fatty acid CoA synthase (ACSL) family of proteins, which are involved in fatty acid degradation, phospholipid remodelling and production of long-chain acyl-CoA esters, could well be involved in this pathway.37 38
Several intracellular bacteria such as Brucella and Legionella interact directly with ER membrane.39 Recently it has been reported that Chlamydia, which is also associated with ReA,40 can also induce ER stress responses for the purposes of exploiting host lipid metabolism.41 Interestingly both Chlamydia and Salmonella have been reported to associate with and/or recruit ER-derived membranes.29 42 Bacterial species such as Salmonella that depend on expansion of membrane compartments to accomodate their growth would benefit from the enhanced lipid production that results from UPR activation. Together, along with observations that HLA-B27 can induce or alter the ER stress environment, UPR induction may be a common feature of intracellular bacteria that reside in vacuoles and may link with the pathology associated with ReA.
Acknowledgments
The authors thank Professor David Holden (Imperial College London), Dr Michael Hensel (Universität Osnabrück) and Dr J Baumler (UC Davis) for the Salmonella strains.
References
Footnotes
DN and SJP are joint senior authors.
Handling editor Josef S Smolen
Contributors All authors contributed to experimental design and to performing the experiments and generating data. All authors contributed to the construction of the manuscript. ANA: contributed to the planning, designing of experiments, interpretation of data and writing of the manuscript. IL: contributed and performed the biochemical analysis of the respective cell lines employed throughout the study, and contributed to the writing of the manuscript. JK-V: contributed to planning, designing and interpretation of the data collated by the microscopic screening, and contributed to the writing of the manuscript. TI: contributed to the design and the use of the UPR reporters. MT: contributed to the microscopic analysis. KM: contributed to the cellular and biochemical analysis of the cell lines employed throughout the study. SA: contributed to the design and generation of the constructs used in the study. NB: contributed to the cellular analysis of the cell lines employed throughout the study. PB: contributed to the cellular and biochemical analyses of the cell lines employed throughout the study and contributed to the writing of the manuscript. MB-E: contributed to the experimental design, data interpretation and writing of the manuscript. KG: contributed to the generation of the constructs and cell lines used throughout the study, data interpretation and writing of the manuscript. DN: contributed to the generation of the constructs and cell lines used throughout the study, data interpretation, experimental design and writing of the manuscript. SJP: contributed to the data interpretation, experimental design and writing of the manuscript, and contributed to the biochemical analysis of the cell lines employed throughout the study.
Funding ANA was funded by ARUK Fellowships Non-Clinical Career DevelopmentFellowship (ref no: 18440). IL was funded by an ARUK PhD studentship (ref no: 17868). ANA and SJP were also in part funded by ARUK (grant 21261).
Competing interests None declared.
Patient consent Not required.
Ethics approval All experiments and procedures were performed as approved by the local ethics.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data are available on request to antony.antoniou@northumbria.ac.uk.