Article Text

Abstract
Objectives To evaluate the efficacy and safety of risankizumab, a humanised monoclonal antibody targeting the p19 subunit of interleukin-23 (IL-23), in patients with active ankylosing spondylitis (AS).
Methods A total of 159 patients with biological-naïve AS, with active disease (Bath Ankylosing Spondylitis Disease Activity Index score of ≥4), were randomised (1:1:1:1) to risankizumab (18 mg single dose, 90 mg or 180 mg at day 1 and weeks 8, 16 and 24) or placebo over a 24-week blinded period. The primary outcome was a 40% improvement in Assessment in Spondylo Arthritis International Society (ASAS40) at week 12. Safety was assessed in patients who received at least one dose of study drug.
Results At week 12, ASAS40 response rates were 25.5%, 20.5% and 15.0% in the 18 mg, 90 mg and 180 mg risankizumab groups, respectively, compared with 17.5% in the placebo group. The estimated difference in proportion between the 180 mg risankizumab and placebo groups (primary endpoint) was –2.5% (95% CI –21.8 to 17.0; p=0.42). Rates of adverse events were similar in all treatment groups.
Conclusions Treatment with risankizumab did not meet the study primary endpoint and showed no evidence of clinically meaningful improvements compared with placebo in patients with active AS, suggesting that IL-23 may not be a relevant driver of disease pathogenesis and symptoms in AS.
Trial registration number NCT02047110; Pre-results.
- ankylosing spondylitis
- DMARDs (biologic)
- treatment
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Ankylosing spondylitis (AS) is a chronic inflammatory rheumatic disease that predominantly affects the axial skeleton, leading to back pain, progressive structural and functional impairment and reduced quality of life.1 AS is generally unresponsive to conventional disease-modifying antirheumatic drugs (DMARDs), and systemic therapy for AS consists of non-steroidal anti-inflammatory drugs (NSAIDs), tumour necrosis factor inhibitors and, more recently, the interleukin (IL) 17A (IL-17A) inhibitor secukinumab.2–4
Several lines of evidence have identified IL-23 as a promising therapeutic target in AS.5 At the genetic level, case–control genome-wide association studies have demonstrated that IL-23 receptor (IL-23R) polymorphisms are associated with an increased risk of developing AS.6 7 In addition, a protective effect of the IL-23RR381Q polymorphism is observed in AS.8 Increased numbers of IL-23-producing cells have been found in facet joints of patients with AS,9 while the number of IL-23-responsive T helper (Th) 22 (Th22), Th17 and gamma/delta T cells are elevated in blood from patients with AS.10 11 Stimulation of peripheral blood mononuclear cells isolated from patients with AS leads to enhanced IL-23 production versus controls.12 Finally, a potential role for the IL-23 pathway in driving entheseal inflammation and bone formation responses in AS has also been highlighted in murine models of spondyloarthritis.13 14 IL-23 is a key driver in the induction and maintenance of Th17 cells.15 The recent approval of the IL-17A inhibitor, secukinumab, for the treatment of AS, supported the clinical hypothesis that direct and specific inhibition of IL-23 would be of therapeutic benefit to patients with AS.2 16 17
Risankizumab (BI 655066/ABBV-066) is a humanised, immunoglobulin G1 monoclonal antibody that selectively inhibits IL-23 by specifically targeting the p19 subunit18 and has shown efficacy in psoriasis, psoriatic arthritis (PsA) and Crohn’s disease.19–22 This proof-of-concept, dose-ranging study assessed the efficacy and safety of risankizumab in patients with active AS.
Methods
Study design
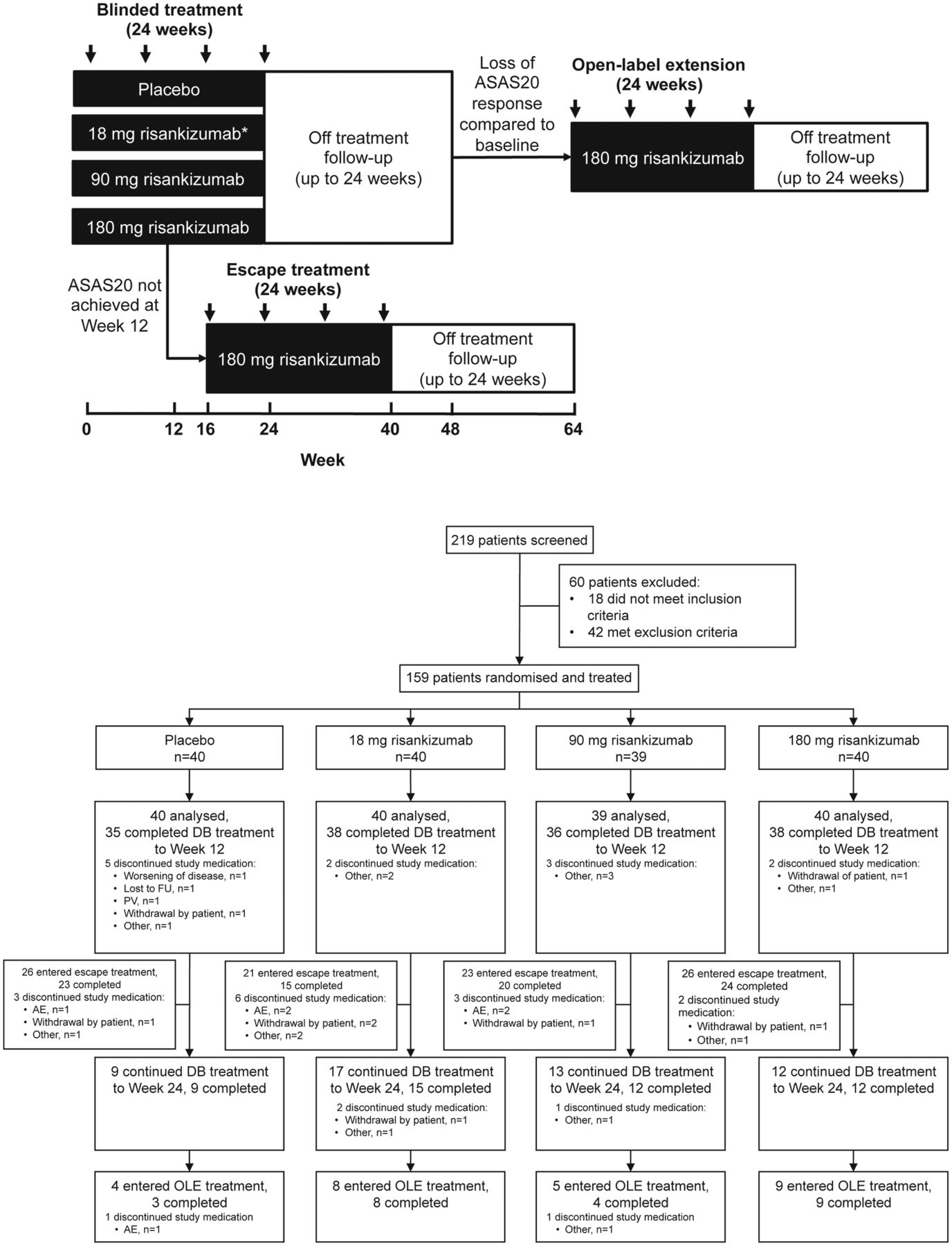
This phase 2, randomised, placebo-controlled, double-blind study was conducted at 47 centres across North America, Europe and East Asia between March and December 2014. Patients were randomly assigned (by interactive response system) in a 1:1:1:1 ratio to one of three regimens of risankizumab (18 mg single dose, 90 mg or 180 mg at day 1 and weeks 8, 16 and 24) or placebo (figure 1A). The doses selected were informed by a phase 1 study in psoriasis and included a 10-fold dose range of risankizumab with a single administration at the low dose (18 mg) that was expected to be subtherapeutic.19 The study comprised a 24-week blinded treatment period, a potential escape treatment period from week 16 up to week 40 and a 24-week open-label extension period (not reported due to small sample size). Each treatment period had a 24-week follow-up (figure 1B). At week 16, escape treatment with 180 mg risankizumab was available for patients not achieving a 20% improvement in Assessment in SpondyloArthritis International Society (ASAS20) at week 12.

Overview of study design and patient disposition. Overview of treatment and observation periods including escape and open-label extension phases (panel A); patients were randomised 1:1:1:1 to one of three regimens of risankizumab (18 mg single dose, 90 mg or 180 mg at day 1 and weeks 8, 16 and 24) or placebo; patients without ASAS20 response at week 12 received escape treatment; patients with a flare of disease activity within 24 weeks of the last double-blind treatment entered the open-label extension. Arrows represent treatment administration. *Patients received 18 mg single dose at day 1 followed by placebo at weeks 8, 16 and 24. Trial profile (panel B). AE, adverse event; ASAS20, 20% improvement in Assessment in SpondyloArthritis International Society; DB, double blind; FU, follow-up; OLE, open-label extension; PV, protocol violation.
Amendments to the protocol can be found in the online supplementary materials. All authors approved the manuscript for submission and vouch for completeness of the data and the fidelity of the study to the protocol.
Supplementary file 1
Patients
Patients aged 18–70 years were eligible if they had definite AS (1984 modified New York criteria23 and local X-ray evaluation), active disease defined as a Bath AS Disease Activity Index (BASDAI) score of ≥4,24 including a value ≥4 for overall level of AS neck, back or hip pain, and documented inadequate response (30 days of optimal daily doses with ≥2 NSAIDs) or intolerance to NSAIDs. Patients previously treated with any biological for AS were excluded, and other biologicals were not permitted during the study. From 2 weeks prior to randomisation and up to 12 weeks of treatment, conventional DMARDs, low-dose systemic steroids (equivalent to ≤10 mg prednisolone/day), NSAIDs or analgesics at stable doses were permitted under the direction of the investigator. See online supplementary material for full inclusion/exclusion criteria. All patients provided written informed consent.
Assessments
The primary endpoint was the proportion of patients achieving an ASAS40 response at week 12. The key secondary endpoint was the change from baseline in the assessment of disease activity based on the AS Disease Activity Score-C reactive protein (ASDAS-CRP) at week 12. All secondary and further endpoints are listed in the online supplementary material. Of these, MRI assessments of the spine and sacroiliac (SI) joints, using the SpondyloArthritis Research Consortium of Canada (SPARCC) MRI indices for scoring inflammation in the SI joint25 and spine,26 were performed pretreatment and post-treatment (at week 24 for patients completing 24-week blinded treatment and at week 12 for patients starting escape treatment) within a subset of patients (see online supplementary material for methodology).
Safety endpoints included adverse events (AEs), serious AEs (SAEs), discontinuation of therapy because of AEs, local tolerability, changes in vital signs and physical examination and laboratory assessments.
Additionally, the IL-23/Th17 pathway biomarker β-defensin 2 and bone remodelling biomarkers associated with AS were evaluated in sera pretreatment and post-treatment at baseline and at week 12. Plasma samples for pharmacokinetic (PK) analysis and immunogenicity assessments were collected at prespecified visits (see online supplementary material for methodologies). A post hoc analysis of change from baseline in C reactive protein (CRP) level was also performed.
Statistical analysis
Sample size was determined based on one-sided comparison between the 180 mg risankizumab dose and placebo using Fisher’s exact conditional test, consistent with the one-sided alternate hypothesis of a week 12 ASAS40 response rate of 43% with 180 mg risankizumab and a rate of 13% with placebo; randomisation of 160 patients (40 per study group) was estimated to provide 89% power using a one-sided test of 0.05 significance. The hierarchical inference strategy protects against type 1 error in other comparisons and other endpoints. Analyses were conducted with all randomised patients who received at least one dose of trial medication (full analysis). The Suissa-Shuster unconditional exact test was used to test the difference in the proportion of patients achieving the primary endpoint between treatment groups per intention-to-treat principles. The CI for the difference in the proportion between the treatment groups was obtained by the Clopper-Pearson method. To control the type I error rate, endpoints were tested in a hierarchical fixed sequence, starting with a primary endpoint comparison between the 180 mg risankizumab group versus placebo, with a 5% (one-sided) significance level. Because the primary analysis failed to show superiority of 180 mg risankizumab over placebo, all remaining analyses were exploratory with nominal p values. For any missing week 12 ASAS40 assessment, non-responder imputation was used. Patients receiving prohibited concomitant medication for AS prior to week 12 were considered as treatment failures. Statistical analyses for secondary and further endpoints are described in the online supplementary material.
Results
Study population and patient disposition
Of the 219 patients screened, 159 underwent randomisation: 40 patients were assigned to 18 mg risankizumab, 39 patients to 90 mg risankizumab, 40 patients to 180 mg risankizumab and 40 patients to placebo. Up to week 12, 12 patients discontinued (ie, did not receive an injection at week 8); there was no imbalance in the frequency of premature discontinuation between risankizumab groups. One patient (placebo) discontinued trial medication due to worsening of AS prior to week 12 (figure 1B). A total of 51 patients fulfilled the ASAS20 response criteria at week 12 and continued treatment to week 24, and 96 patients were switched to escape treatment.
Demographic and baseline characteristics were balanced across study groups, with some variation related to the limited sample size (table 1). In particular, a lower number of HLA-B27-positive patients were randomised to the placebo group, most likely reflecting the higher number of Asian patients in the placebo group compared with risankizumab groups. Overall, the baseline disease activity and CRP levels of patients in this study were similar or somewhat less severe to those of patients in recent AS clinical trials.2 16
Baseline demographics and clinical characteristics
Efficacy
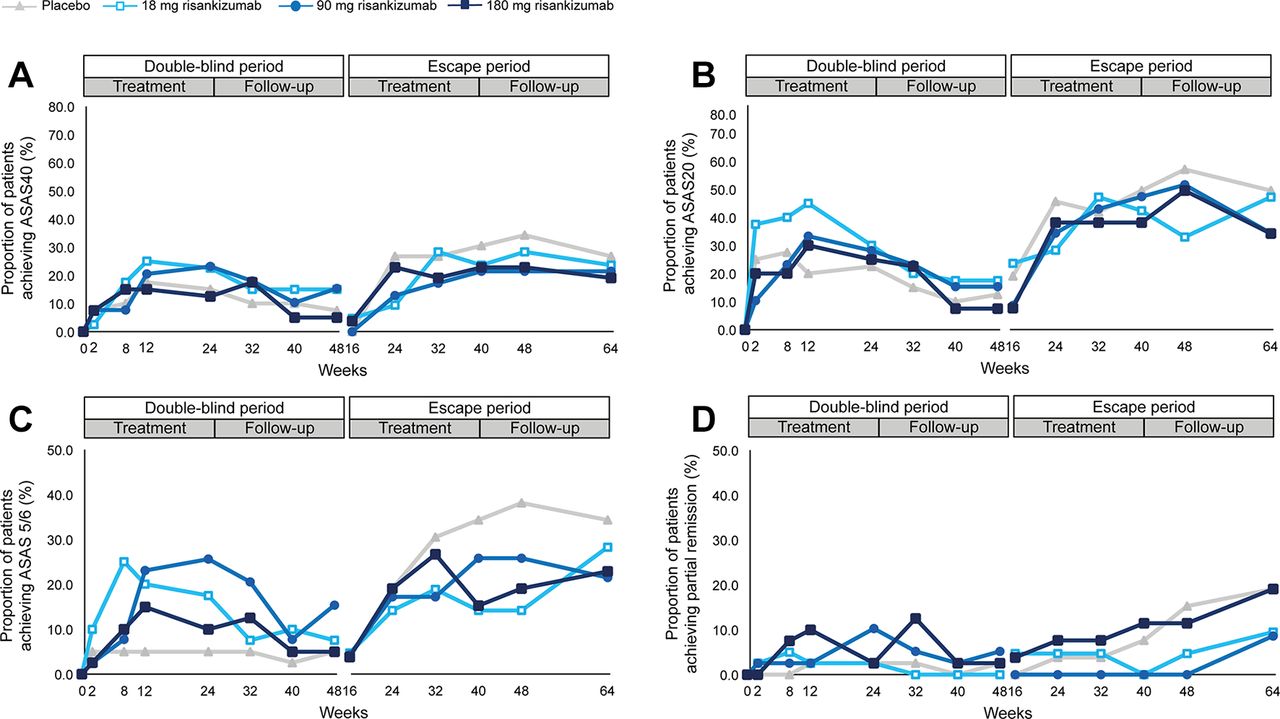
The primary endpoint, ASAS40 response at week 12, was not met. ASAS40 response at week 12 was achieved by 25%, 21% and 15% of patients in the 18 mg, 90 mg and 180 mg risankizumab groups, respectively, compared with 18% in the placebo group. The estimated difference in the proportion of ASAS40 responders between the 180 mg risankizumab group and placebo (primary endpoint) was –2.5% (95% CI –21.8 to 17.0; p=0.42) and 7.5% (95% CI –12.1 to 26.6; p=0.27) and 3.0% (95% CI –15.9 to 20.8; p=0.41) between the 18 mg and 90 mg risankizumab groups versus placebo, respectively (figure 2A). The prolongation of risankizumab treatment for up to 40 weeks by patients receiving escape treatment (180 mg risankizumab) did not substantially improve ASAS40 attainment rates.

Response rates for ASAS40, ASAS20, ASAS 5/6 and ASAS partial remission during double-blind and escape treatment and follow-up periods. Clinical response rates over time for double-blind and escape treatment periods. ASAS40 (panel A), ASAS20 (panel B), ASAS 5/6 (panel C) and partial remission (panel D). NRI was used for missing data. Number of patients entering the double-blind treatment were: placebo: n=40; 18 mg risankizumab: n=40; 90 mg risankizumab: n=39; and 180 mg risankizumab: n=40. Patients entering escape treatment received 180 mg risankizumab; responses shown for the escape period are by the original randomised treatment (placebo: n=26; 18 mg risankizumab: n=21; 90 mg risankizumab: n=23; 180 mg risankizumab: n=26). Values for all data points are provided in online supplementary tables S1–S4. ASAS, Assessment in SpondyloArthritis International Society; NRI, non-responder imputation.
ASAS20 response at week 12 was achieved by 45%, 33% and 30% of patients in the 18 mg, 90 mg and 180 mg risankizumab groups, respectively, compared with 20% in the placebo group (figure 2B). ASAS 5/6 response at week 12 was achieved by 20%, 23% and 15% of patients in the 18 mg, 90 mg and 180 mg risankizumab groups, respectively, compared with 5% in the placebo group (figure 2C). Partial remission (ASAS criteria) at week 12 was achieved by 3%, 3% and 10% of patients in the 18 mg, 90 mg and 180 mg risankizumab groups, respectively, compared with 3% in the placebo group (figure 2D).
Median change (IQRs) from baseline in ASDAS-CRP at week 12 were –0.7 (–1.3 to –0.2), –0.6 (–1.2 to 0.0) and –0.7 (–1.1 to –0.3) for the 18 mg, 90 mg and 180 mg risankizumab groups, respectively, compared with –0.3 (–1.0 to 0.2) for the placebo group (figure 3A). A dose-dependent reduction in CRP was observed with risankizumab compared with placebo at week 12 (figure 3B). For the BASDAI score (figure 3C) and other efficacy endpoints (Bath Ankylosing Spondylitis Functional Index, Bath Ankylosing Spondylitis Metrology Index, tender joint count, swollen joint count and Maastrich Ankylosing Spondylitis Enthesitis Score), there were no meaningful changes over time between the risankizumab groups and placebo (online supplementary table S8).

{kind=link}
{kind=link}
{kind=link}
Change from baseline in ASDAS-CRP, CRP and BASDAI over time to week 12. Change from baseline in ASDAS-CRP (panel A), CRP (mg/L) (panel B) and BASDAI (panel C) over time to week 12. Median (IQR) changes are shown (observed). The values under each plot are the number of patients per treatment arm with a valid measurement at the specified time point. *P=0.0229 and p=0.0101 for median change in ASDAS-CRP for 18 mg and 180 mg risankizumab, respectively, versus placebo at week 12. Values for all data points are provided in online supplementary tables S5–S7. ASDAS-CRP, Ankylosing Spondylitis Disease Activity Score-CRP; BASDAI, Bath Ankylosing Spondylitis Disease Activity Score; CRP, C reactive protein.
Only ASAS20 responders at week 12 (32%) continued blinded treatment up to week 24 (94% completing blinded treatment; figure 1), and most ASAS40 responders at week 12 (n=31) remained responders up to week 24 (n=29).
Subgroup analyses of ASAS40 response at week 12 by baseline CRP level, geographic region or morning stiffness did not reveal a higher ASAS40 response for any risankizumab group compared with placebo (online supplementary table S9).
Findings from the MRI analysis were generally consistent with clinical effects (online supplementary table S10 and figure S1). In patients who continued treatment to week 24, risankizumab had no effect on SPARCC SI joint score compared with placebo (online supplementary table S10) but improved SPARCC total spine (median (IQR)) change from baseline in the 90 mg (–6.0 (–12.0 to –2.8); p=0.0046) and 180 mg (–3.8 (–8.0 to 1.3); p<0.05) risankizumab groups, compared with placebo at week 24. For patients who switched to escape treatment, there was no difference in change in SPARCC total spine or SI joint scores at week 12 or from weeks 12–24 between treatment groups (online supplementary table S10).
There were minimal differences between the changes from baseline in levels of β-defensin 2 and biomarkers of bone remodelling (dikkopf-1, sclerostin, BMP-7 and osteocalcin) in patients receiving risankizumab versus placebo over time. A heat map showing the percentage change from baseline to week 12 in biomarkers versus change in clinical and MRI parameters in ASAS20 responders and non-responders is provided in online supplementary figure 2.
Safety
Up to week 16, the rate of AEs was comparable between the risankizumab groups and the placebo group. Most reported AEs were of mild or moderate intensity, and there were no reports of severe infections across all groups. Three patients in the placebo group had AEs leading to the premature discontinuation of trial medication. The frequency of investigator-defined drug-related AEs was low and comparable between the risankizumab and placebo groups (table 2). The most common AEs reported with risankizumab and/or placebo included nasopharyngitis, headache and fatigue. Up to the end of the trial, the rate and profile of AEs in the risankizumab total group was consistent with the up to 16 weeks data in this trial and with other risankizumab trials (online supplementary table S11). All SAEs were deemed non-drug related by the investigator and were graded as serious due to the need for hospitalisation. Local tolerability issues at the injection site were uncommon and mostly mild, and no anaphylactic reactions or inflammatory bowel syndrome diagnoses were reported.
AEs summary up to 16 weeks
Overall, PK parameters and the exposure (online supplementary table S12) of risankizumab in patients with AS were comparable with patients with psoriasis treated with risankizumab in prior trials.19 20 Antidrug antibody (ADA) incidence was 14%, with 4% testing positive at baseline before treatment (pre-existing ADA) and only 3% testing positive in the neutralising antibody assay. In most patients, ADAs were transient and of low titre (≤16). These results were similar to those reported in patients with psoriasis in a previous trial.20 ADAs did not impact the exposure or safety of risankizumab.
Discussion
AS is a complex polygenic disease showing a genetic association with the IL-23 pathway.6 8 However, in this phase 2, proof-of-concept study, a significant improvement in the proportion of patients achieving an ASAS40 response with risankizumab at week 12, compared with placebo, was not demonstrated. Longer treatment (up to 40 weeks, by patients receiving escape treatment with 180 mg risankizumab) did not substantially improve ASAS40 attainment rates. The lack of efficacy was also confirmed by most secondary and other endpoints. While modest reductions of ASDAS-CRP were evident with risankizumab, a clinically relevant change of ≥1.1 was not achieved.27 ASAS 5/6 at week 12 was the only secondary endpoint to demonstrate a higher response rate in each of the risankizumab treatment groups compared with placebo. The primary driver in both ASDAS-CRP and ASAS 5/6 reductions was CRP, with minimal changes in other composite endpoint parameters. Findings from MRI analysis indicated a limited impact of risankizumab at week 24 on SPARCC total spine score in patients with a clinical response. Despite this, trial discontinuations due to lack of efficacy were low.
Risankizumab was generally well tolerated in this study, and there were no new or unexpected safety signals identified.19–22
Findings of this study are in contrast to those reported for secukinumab, an IL-17A inhibitor that demonstrated efficacy in patients with AS.2 16 17 In an open-label trial, ustekinumab (an IL-12/23 inhibitor) showed efficacy in 20 patients with AS through 28 weeks; however, recently, phase 3 trials in AS and non-radiographic axial spondyloarthritis were terminated for not meeting key efficacy endpoints (ClinicalTrials.gov identifiers: NCT02438787 and NCT02407223).28 The failure of risankizumab in this study was unexpected, and the underlying reasons remain unclear. Theoretically, it is possible that risankizumab may not have sufficiently blocked levels of IL-23 in the target organ (ie, the spine and SI joints). However, this is unlikely for the following reasons: risankizumab has shown proof-of-concept in psoriasis,19 20 PsA22 and Crohn’s disease;21 the observed PK exposure levels were consistent with effective exposures in psoriasis; risankizumab had a dose-dependent effect on CRP; and that an evident improvement in SPARCC spine score in the 90 mg and 180 mg treatment groups was seen in patients who had remained within 24-week blinded treatment in this study. Enrolment criteria in this study excluded patients with concomitant inflammatory bowel disease or PsA; however, patients with concomitant psoriasis were permitted, but unfortunately information on this comorbidity was not collected systematically, and thus we were unable to assess whether there were any improvements in psoriatic lesions, as evidenced in previous studies of risankizumab in patients with psoriasis.
Alternatively, risankizumab may not have sufficiently blocked all relevant sources of IL-17 production. Previously, risankizumab has been shown to reduce the levels of IL-17A, IL-23A and associated transcripts in lesional skin of patients with psoriasis.19 Although it is well characterised that one function of IL-23 is the induction and maintenance of Th17 cells,15 it is likely that IL-23-independent sources of IL-17 are still active, such as IL-17-secreting mast cells and mucosal-associated invariant T cells.29 In addition, in in vivo autoimmune mouse models, dual inhibition of IL-17 and IL-23 was more efficacious in reducing disease than targeting either cytokine alone,30 confirming that the relationship between IL-23 and IL-17 is not linear. Discordant effects of IL-17 and IL-23 inhibition were previously observed in Crohn’s disease, where a lack of clinical efficacy of IL-17 inhibitors and even disease exacerbation and elevated inflammatory markers (including serum CRP and faecal calprotectin) were reported,15 whereas IL-23 inhibition has shown proof-of-concept in Crohn’s disease.21
The results of this study challenge the notion that IL-23 is a relevant driver of AS disease pathogenesis and symptoms. This is further supported by analyses showing no overall differences in change in levels of an antimicrobial peptide and IL-23/Th17 pathway biomarker and select biomarkers of bone remodelling in patients receiving risankizumab versus placebo. The role of changes in β-defensin 2 in disease processes occurring in AS is less understood compared with psoriasis,20 21 but the results from the current study suggest there may be differences between IL-23/Th17 pathway biomarkers associated with psoriasis and those associated with AS.
A possible limitation of this study is that the design did not include a loading dose, as has been included in some clinical studies of biological agents in AS and in phase 2 studies for risankizumab in psoriasis (treatment at weeks 0, 4 and 16)20 and Crohn’s disease (induction dosing every 4 weeks intravenously for 12 weeks);21 therefore, the study design might have potentially contributed to the negative results. However, because there was no convincing evidence for a dose response in this study, and a single dose of 18 mg risankizumab has demonstrated efficacy in a psoriasis phase 2 trial,20 it is unlikely that a loading dose would have improved the treatment response in AS. Studies with secukinumab have also indicated that a loading dose is not required, thus leading it to be optional in the secukinumab label.2 31 Another limitation of the study design was that there was no comparator arm in the escape group that received four additional doses of open-label 180 mg risankizumab, making the efficacy data for the 96 patients in this group difficult to interpret.
In this proof-of-concept study, risankizumab was not effective in reducing the signs and symptoms of AS. The lack of efficacy of this IL-23 inhibitor at subcutaneous doses previously shown to be highly effective in psoriasis suggests that, despite a genetic association with the IL-23 pathway, IL-23 may not be a relevant driver of disease pathogenesis and symptoms in AS.
Acknowledgments
We would like to thank all the patients and study investigators who participated in the clinical study described here. Additional thanks to Kelly Coble for expertise in PK analysis, Oliver Kleiner for expertise in biomarker analyses, Yu-Wei Chang for statistical support and Richard Vinisko for PK and biomarker analyses.
References
Footnotes
Handling editor Josef S Smolen
Contributors DB, DBH, MØ, SJP and PS contributed to study design. DB, PJ, T-HK, MØ, AS, CS, JS, L-ST and JC-CW contributed to data collection. All authors had full access to the study data, contributed to data analysis, data interpretation, writing and review of the manuscript and approved the final version for publication.
Funding This study was funded by Boehringer Ingelheim. Editorial assistance in the development of this manuscript was provided by Leigh Church of SuccinctChoice Medical Communications (London, UK), funded by Boehringer Ingelheim.
Competing interests DB reports grants from AbbVie, Pfizer, UCB, MSD, Novartis and Eli Lilly and part-time employment at UCB. MØ reports grants, personal fees and non-financial support from AbbVie, BMS, Merck, UCB and Novartis; grants and personal fees from Celgene; personal fees and non-financial support from Janssen, Pfizer and Roche; and personal fees from Boehringer Ingelheim, Eli Lilly, Sanofi, Regeneron, Orion and Hospira. JS reports personal fees from Boehringer Ingelheim, AbbVie, Janssen, Lilly, Merck, Novartis, Pfizer and UCB. PJ reports grants from AbbVie, Daiichi Sankyo, Boehringer Ingelheim, Lilly, Novartis, Roche and UCB and grants and personal fees from BMS and Pfizer. YD, CP, SV, DBH, SA, PS and SJP report being employees of Boehringer Ingelheim.
Patient consent Not required.
Ethics approval The study protocol was approved by the institutional review board or ethics committee at each participating centre. The study was conducted according to the Declaration of Helsinki and the International Conference on Harmonisation guidelines. Safety data were periodically evaluated by an independent data monitoring committee. All patients provided written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The authors confirm that the data supporting the findings of this study are available within the article or its supplementary materials.