Article Text

Abstract
Objective To demonstrate clinical equivalence of adalimumab biosimilar candidate BI 695501 with Humira.
Methods Patients with active rheumatoid arthritis on stable methotrexate were randomised to BI 695501 or Humira in a double-blind, parallel-group, equivalence study. At week 24, patients were rerandomised to continue BI 695501 or Humira, or switch from Humira to BI 695501. The coprimary endpoints were the percentage of patients achieving the American College of Rheumatology 20% response criteria (ACR20) at weeks 12 and 24. Further efficacy and safety endpoints and immunogenicity were assessed up to week 58.
Results 645 patients were randomised. At week 12, 67.0% and 61.1% (90% CI –0.9 to 12.7) of patients receiving BI 695501 (n=324) and Humira (n=321), respectively, achieved ACR20; at week 24 the corresponding values were 69.0% and 64.5% (95% CI –3.4 to 12.5). These differences were within prespecified margins (week 12: 90% CI (–12% to 15%); week 24: 95% CI (−15% to 15%)), demonstrating therapeutic bioequivalence. 593 patients were rerandomised at week 24. Up to week 48, mean change from baseline in Disease Activity Score 28-erythrocyte sedimentation rate and ACR20/ACR50/ACR70 response rates were similar across the switched (n=147), continuous BI 695501 (n=298) and continuous Humira (n=148) groups. Similar immunogenicity (antidrug antibodies (ADAs), ADA titres and neutralising antibodies) was seen between BI 695501 and Humira (to week 24) and across rerandomised groups (to week 48). Safety and tolerability profiles were similar between groups.
Conclusions BI 695501 demonstrated similar efficacy, safety and immunogenicity to Humira; switch from Humira to BI 695501 had no impact on efficacy, safety and immunogenicity.
Trial registration number NCT02137226, Results.
- rheumatoid arthritis
- anti-tnf
- autoimmune diseases
- dmards (biologic)
- treatment
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Biosimilars are reproductions of existing biologic molecules that have a high degree of similarity to their reference products, including their molecular structure, biological function and effect in patients, that is, efficacy, safety and immunogenicity. Development programmes for biosimilars are specifically designed to demonstrate similarity to the reference product1; they do not assess efficacy and safety profiles versus a current standard of care. These requirements, defined by the Food and Drug Administration (FDA)1 and European Medicines Agency (EMA),2 include a phase III clinical trial comparing clinical efficacy and safety of the biosimilar with its reference product in a clinical model that is sensitive to detect any potential clinically meaningful differences between the two versions of the molecule.1 3
The wide use of biologics across a number of diseases has led to significant improvements in patients’ health. This has come with an increase in healthcare expenditure.4 However, the advent of biosimilars to infliximab, etanercept and rituximab has introduced more treatment choice5 and led to cost reductions.
The tumour necrosis factor inhibitor Humira (adalimumab, AbbVie) is an established biologic treatment for a number of immune-mediated inflammatory diseases, including rheumatoid arthritis (RA), psoriatic arthritis and inflammatory bowel disease. A number of biosimilar candidates to Humira are currently in development, including the recently approved BI 695501 (Cyltezo, adalimumab-adbm, Boehringer Ingelheim).6 Extensive comparison of the physicochemical structure and biologic function of BI 695501 and Humira showed structural similarity and comparable functionality (Sonderegger I, Wittner M, 2018. Manuscripts in preparation). Furthermore, the VOLTAIRE-PK study (NCT02045979) established three-way pharmacokinetic similarity between BI 695501, and European Union (EU)-approved and USA-approved Humira.6
The VOLTAIRE-RA trial constituted the final step of the biosimilarity assessment for BI 695501.
Methods
Study design
VOLTAIRE-RA was a randomised, double-blind, parallel-arm, 58-week equivalence trial of BI 695501 and USA-sourced Humira (NCT02137226; figure 1A) in 14 countries (115 sites). Patients with moderate-to-severe RA on stable methotrexate (MTX) were randomised 1:1 to receive BI 695501 or Humira 40 mg subcutaneously by prefilled syringe once every 2 weeks for 24 weeks by suitably qualified, designated blinded trial personnel either on-site or at the patient’s home. First doses of trial medication were administered at the site. Randomisation (via an interactive response technology system; Almac Clinical Technologies, Souderton, Pennsylvania, USA) included stratification according to region (Asia, EU, Latin America, USA) and prior exposure to a biologic agent (yes/no) (see online supplementary appendix A for further details). Patients originally randomised to Humira were rerandomised at week 24 to either continue Humira (continuous Humira) or transition to BI 695501 (Humira to BI 695501). Patients originally randomised to BI 695501 were dummy-rerandomised to continue BI 695501 (continuous BI 695501). Rerandomisation was stratified by prior exposure to a biologic agent only.
Supplemental material

VOLTAIRE-RA study design (A) and patient disposition (B). *Patients continued with methotrexate 15–25 mg/week. Methotrexate 10–14 mg/week was permitted for patients with documented intolerance to higher doses of methotrexate. †Humira 40 mg/0.8 mL solution for subcutaneous injection. EOT, end of treatment; EOW, every other week; n, number of patients per group.
At the end of the trial, qualifying patients could enter an open-label extension (OLE; VOLTAIRE-RAext; NCT02640612), where all patients received BI 695501 for ≤48 weeks; otherwise, safety follow-up occurred at week 58. The study was conducted in accordance with the Declaration of Helsinki. All patients provided written informed consent.
Objectives and endpoints
The primary objective of the study was to demonstrate equivalent clinical efficacy of BI 695501 and Humira. Coprimary endpoints were the percentage of patients achieving the American College of Rheumatology 20% response criteria (ACR20) at week 12 (requested by FDA) and week 24 (requested by EMA). Prespecified secondary endpoints were change from baseline in Disease Activity Score in 28 joints (DAS28) using erythrocyte sedimentation rate (ESR) at weeks 12 and 24, and the percentage of patients with drug-related treatment-emergent adverse events (AEs). Further endpoints included ACR-based and DAS-based parameters at various time points, quality of life (36-Item Short-Form Health Survey (SF-36) V.2), AEs (including infections/serious infections, hypersensitivity reactions, drug-induced liver injury, injection site reactions) and immunogenicity (antidrug antibodies (ADAs), neutralising antibodies (nAbs), drug levels).
Patients
Adults (18–80 years) with moderately to severely active RA for ≥6 months, defined by ≥6 swollen joints (66 joint count) and ≥6 tender joints (68 joint count), at screening and baseline, and either ESR >28 mm/hour or C reactive protein (CRP) >1.0 mg/dL at screening, were enrolled. Patients must have received 15–25 mg/week MTX background treatment for ≥12 weeks prior to enrolment. MTX 10–14 mg/week was permitted for patients intolerant to higher doses. Patients could have been on oral corticosteroids ≤10 mg/day prednisolone or equivalent (stable for 4 weeks prior to day 1) and stable non-steroidal anti-inflammatory drugs for 2 weeks prior to day 1.
Exclusion criteria included previous RA treatment with adalimumab or >1 other biologic, active infection, hypersensitivity reactions or AEs to agents similar to the study drugs or their excipients (full criteria available in online supplementary appendix B).
Statistical analyses
For determination of the primary endpoint, non-responder imputation was used for patients who discontinued prior to that time point. For patients who had not discontinued but had missing data, multiple imputation was used. At each time point (weeks 12 and 24), and on each of the complete data sets following the imputation, logistic regression was applied, including fixed, categorical effects of treatment and prior exposure to a biologic agent (yes/no), and continuous effects of baseline DAS28-ESR. The multiple risk differences and CIs on the individual complete data sets were calculated using the Reeve method,7 and combined using Rubin’s rules.8
Region was not included in the model due to sparse data in some regions. This was known shortly after final recruitment and included in a protocol amendment prior to database lock.
The primary endpoint, analysed as described above and based on the full analysis set (FAS), was met if the upper and lower CIs of both coprimary endpoints were contained within the prespecified margins. Equivalence was achieved when the difference in ACR20 response rates (BI 695501 minus Humira) was within −12% and 15% (90% CI; week 12 per FDA consultation) and within −15% and 15% (95% CI; week 24 per EMA consultation). An FDA-agreed asymmetrical margin at week 12 was defined, with a slightly higher upper bound of +15% to allow for variations in techniques and response rates used in the calculation of the margins. For this test to be performed with adequate power (86%–91%), a sample size of ~650 patients was needed (FAS). This sample size was based on an assumed treatment difference in ACR20 response rates of 0%, a standard proportion of 59% and an asymmetrical equivalence margin of (−12% to 15%) at week 12, with corresponding values of 0%, 63% (−15% to 15%) at week 24.
The FAS contained all patients who received at least one dose of trial drug and who had all measures required for the efficacy endpoints (ACR20 at weeks 12 and 24) at baseline and at least once postbaseline. The per-protocol analysis set (PPS) contained all patients in the FAS who did not experience any important protocol deviations relevant for efficacy (eg, severe deviation to the restricted disease-modifying anti-rheumatic drug (DMARD) therapy prior to primary endpoint assessment). The safety analysis set (SAF) contained all patients who received at least one dose of trial drug. Descriptive safety data were coded according to MedDRA V.19.0. Data were analysed using SAS software Version 5.0.
The secondary efficacy endpoint of change from baseline in DAS28-ESR was assessed via an analysis of covariance model, using multiple imputation method for missing data. ACR20 at week 48, ACR50 and ACR70 and further efficacy endpoints were computed using the same missing data methodology. Exploratory endpoints were analysed by descriptive statistical methods.
Immunogenicity
Immunogenicity evaluations were performed (SAF) as previously described (overview available in online supplementary appendix C).6
Results
Patient disposition and baseline characteristics
The first patient was screened on 4 February 2015. Across 137 centres, 645 patients were randomised (3 March 2015–18 October 2015) 1:1 to BI 695501 (n=324) and Humira (n=321) (SAF). Six patients were excluded from the FAS (lack of postbaseline efficacy assessment); 38 patients were excluded from the PPS (protocol deviations). At week 24, 593 patients were rerandomised to continuous BI 695501 (n=298), continuous Humira (n=148) and Humira to BI 695501 (n=147); 85 (13.2%) patients discontinued the trial prematurely. Last patient, last visit occurred on 18 October 2016. There were no differences in the rate of treatment or trial discontinuation between treatment groups. Patient disposition and geographical distribution are presented in figure 1B and online supplementary table S1, respectively.
Baseline demographics and clinical characteristics were balanced between treatment groups (table 1).
Baseline demographics and clinical characteristics (SAF)
Efficacy
Results at week 24
Both coprimary endpoints met the predefined criteria, demonstrating therapeutic equivalence of BI 695501 and Humira at weeks 12 and 24 (table 2). The difference in the proportion of patients achieving an ACR20 response was within the prespecified interval at week 12 (90% CI −0.9 to 12.7) and week 24 (95% CI −3.4 to 12.5). Primary and sensitivity analyses of the coprimary endpoints are presented in online supplementary figure S1. A post-hoc analysis to determine relative risk is presented in online supplementary table S2.
Primary efficacy endpoint: estimate and CIs for differences in ACR20 response rate at week 12 and week 24 (FAS)
As a sensitivity analysis, the primary efficacy analysis was repeated on the PPS (same imputation methodology; online supplementary figure S1). The similarity of ACR20 responses in the two groups at weeks 12 and 24 was independent of baseline demographic and clinical characteristics (online supplementary figure S2). The analysis of the secondary efficacy endpoints supported the findings of the primary efficacy analysis. The mean percentage of patients meeting the ACR20/50/70 response criteria was similar in each treatment group at weeks 12 and 24 (figure 2A). The mean change from baseline in DAS28-ESR was similar between the two treatment groups at weeks 12 and 24 (online supplementary table S3; figure 2B).

Week 24 results (A–C). Percentage of patients with ACR20/ACR50/ACR70 responses; bars show SEs (A). Mean DAS28-ESR; bars show SDs (B). EULAR responses (C). Week 48 results (D–F). Percentage of patients with ACR20/ACR50/ACR70 responses. Bars show SEs (D). Mean DAS28-ESR; bars show SDs (E). EULAR responses (F). ACR, American College of Rheumatology; DAS28-ESR, Disease Activity Score in 28 joints-erythrocyte sedimentation rate; EULAR, European League Against Rheumatism.
The percentage of patients with European League Against Rheumatism (EULAR)-defined ‘good’ and ‘moderate’ response rates was similar at weeks 12, 24 and 48 (figure 2C; ACR/EULAR Boolean definition9 of remission available in online supplementary table S4). Both treatment groups showed a similar increase in the SF-36 physical and mental component scores (weeks 12 and 24), indicating a similar improvement in quality of life (online supplementary figure S3).
Results at week 48
Results from baseline to week 48 are presented as per the three treatment groups generated at rerandomisation (week 24). ACR20/50/70 response rates and mean change from baseline in DAS28-ESR were similar across the switched and the continuous groups (figure 2D,E). Similar percentages of patients had ‘good’ and ‘moderate’ EULAR response rates at week 48 in each group (figure 2F).
Safety
Safety follow-up was to week 58 for all patients who did not enter the OLE. The proportion of patients with drug-related AEs was similar between the treatment groups. Overall, safety findings were similar between the continuous BI 695501 and Humira arms from day 1 to week 58, and between the rerandomised groups from week 24 to 58 (table 3). The frequency of AEs with an incidence of ≥3% to week 58 is presented in online supplementary table S5. Among serious AEs, infections and infestations was the most common system organ class (0.6% for BI 695501 vs 4.0% for Humira). No deaths were reported during the study.
Overview of AEs (SAF)
The most frequently reported AEs leading to drug discontinuation were acute pyelonephritis (n=2) and urticaria (n=2) (both in the Humira group only). Up to week 24, serious infections were pneumonia (n=4), acute pyelonephritis (n=2), and appendicitis, infective arthritis and bronchitis (each, n=1) (Humira group only). Cellulitis was reported for one patient (BI 695501 group). From week 24 to week 58, serious infections were pneumonia in one patient (continuous Humira group), and influenza, viral pneumonia and salmonella sepsis in one patient (Humira to BI 695501 group).
Immunogenicity
Immunogenicity data were available at week 24 (SAF population; 92.2%; n=595/645) and week 48 (87.8% of total randomised; n=566). At baseline, 32 (5.0%) patients had ADAs against adalimumab (BI 695501 group, n=11; Humira group, n=21). In 25/32 patients these ADAs were neutralising (9 BI 695501; 16 Humira).
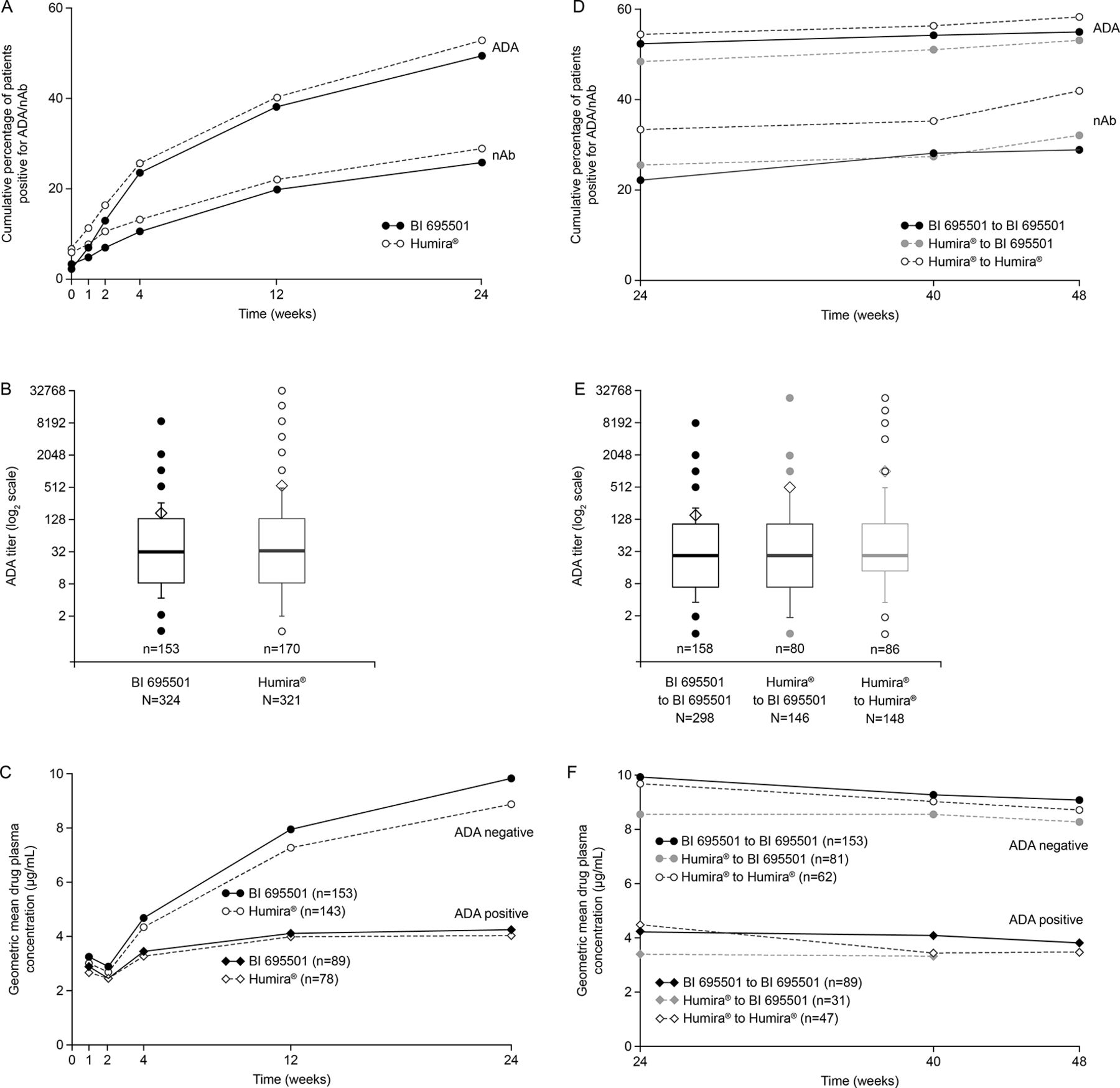
Overall 50.2% of the patients were ADA-positive at any time point up to week 24. The ADA frequencies up to week 24 were similar in the BI 695501 (47.4%) and in the Humira groups (53.0%) (figure 3A). ADA titres at week 24 (figure 3B) and nAb frequencies up to week 24 (figure 3A) were also similar between the groups. Whether or not patients transitioned from Humira to BI 695501 or continued on Humira did not influence subsequent ADA frequency and titres. Similar immunogenicity was observed after week 24 in all rerandomised groups (ADA-positive patients at any time point up to week 48, figure 3D; nAb-positive patients at any time point up to week 48, figure 3D; ADA titres at week 48, figure 3E).
{kind=link}
{kind=link}
{kind=link}
Week 24 results (A–C). Percentage of patients with positive ADA/nAb test (A). ADA titre (B). Drug plasma concentration by presence of ADAs (C). Week 48 results (D–F). Percentage of patients with positive ADA/nAb test (D). ADA titre (E). Drug plasma concentration by presence of ADAs (F). ADA, antidrug antibodies; n, number of patients per group; nAb, neutralising antibodies.
The impact of ADA on drug plasma levels at weeks 24 and 48 is shown in figure 3C,F. A lower drug concentration was measured in ADA-positive patients compared with ADA-negative patients. This effect was independent of treatment group. Overall, in a post-hoc exploratory analysis, ADA-positive patients had a numerically slightly lower median change from baseline in DAS28-CRP at week 48 than ADA-negative patients (week 48: ADA-positive −2.25, ADA-negative −2.52).
Discussion
The efficacy, safety and immunogenicity results of this study demonstrate clinical equivalence of BI 695501 and Humira. In combination with the phase I VOLTAIRE-PK study data6 plus prior physicochemical and functional analyses (manuscripts in preparation), VOLTAIRE-RA completes the similarity assessment of the adalimumab biosimilar BI 695501.
Selection of study design and endpoints
A biosimilar phase III study needs to be designed to optimise the chance of detecting potential clinical differences between the biosimilar candidate and reference product.1 2 A suitable clinical model is characterised by a combination of certain factors, including a disease and a population that will respond to the treatment with a large effect size and endpoints that are sensitive to measure those responses. A homogeneous population increases the sensitivity to detect differences because of the reduction of interindividual variability. The clinical model is usually selected in consultation with regulatory agencies.
Adalimumab is a standard of care in a wide range of autoimmune diseases and is commonly used in adults and children. Phase III studies for other adalimumab biosimilar candidate molecules have been completed or are ongoing in adult patients with RA (eg, with SB5,10 FKB32711 and PF-0641029312) or plaque psoriasis (including with MYL-1401A,13 CHS-1420,14 M92315 and MSB1102216) or both (ABP 50117 18 and GP201719 20). RA was selected for the main phase III study with BI 695501 due to its sensitivity to adalimumab, the availability of ACR20 as a well-established and sensitive measure of disease activity reduction, and operational feasibility. Supportive phase III studies are ongoing with BI 695501 in plaque psoriasis (NCT02850965) and Crohn’s disease (NCT02871635).
Switching from reference product Humira to BI 695501
When switching from the reference product to a biosimilar for which clinical similarity has been established (as presented here for Humira to BI 695501), one would not expect a change in efficacy or safety, although natural fluctuation of disease activity in individual patients may occur. It is important that such treatment switches are studied and understood as this may become common practice within routine care (eg, many patients transitioned from Remicade to its biosimilars as they became available).
Here, patients who had been treated with Humira (baseline–24 weeks) were randomised to either continue Humira or switch to BI 695501 (weeks 25–48). No differences were detected between these two groups with regard to adherence to treatment, efficacy, safety and immunogenicity. Future, observational studies could help confirm these findings in the real-world setting. Since biosimilars to Remicade have become available, a number of observational and interventional studies (such as the NOR-SWITCH study)21 have confirmed the findings of the development programme of CT-P13 (Remsima, Inflectra), suggesting that developing biosimilars in a programme that relies on analytical, preclinical and limited clinical studies is a robust concept.
Overall assessment of efficacy and safety
The efficacy data from VOLTAIRE-RA indicate that BI 695501 and Humira have therapeutic equivalence at week 12 and week 24. Up to week 48, the mean change from baseline in DAS28-ESR and ACR20/50/70 response rates was similar across the switched and continuous groups. BI 695501 and Humira demonstrated similar safety and tolerability; there were no new safety findings for adalimumab. The frequency of hypersensitivity or injection site reactions was similarly frequent in all treatment groups.
Overall assessment of immunogenicity
Immunogenicity is a key aspect of the clinical similarity evaluation of a biosimilar agent. Therefore, a highly sensitive and drug-tolerant ADA assay was developed and applied during clinical development of BI 695501. Different and often less sensitive ADA and nAb assays were used in historical trials (eg, pivotal trials with Humira22). This can explain previous reports of different frequencies of ADA-positive and nAb-positive patients detected in earlier studies. Overall, similar immunogenicity (ADA frequency and titres, and nAb frequency) was observed between BI 695501 and Humira throughout this study. Patients switching from Humira to BI 695501 did not demonstrate increased immunogenicity or more hypersensitivity reactions compared with patients continuing to receive Humira.
As expected, an inverse correlation between ADAs and drug plasma concentration was detected. This effect was similar between the BI 695501 and Humira at week 24 and between the three study groups at week 48. This confirms previous data from the VOLTAIRE-PK study6 showing a comparable impact of ADA on key pharmacokinetic parameters for BI 695501, and USA-approved and EU-approved Humira. An explanation of the pre-existence of antibodies in otherwise drug-naïve subjects is given in online supplementary appendix D.
Adalimumab biosimilar landscape
The introduction of adalimumab was a major step forward for patients suffering from certain chronic immune-mediated diseases. Its benefits for patients, along with the prevalence of its indications, led to healthcare system costs exceeding US$15 billion (2016). It is therefore unsurprising that several companies are developing biosimilars to Humira. Currently, Amgen’s ABP 501 (Amgevita/Amjevita) is FDA-approved and EMA-approved.22 23 Clinical study results are typically similar for different biosimilar candidates due to comparable study designs (regulator agency requirement) and inclusion of AbbVie’s reference product Humira as the common comparator.
Conclusion
VOLTAIRE-RA showed that BI 695501 and Humira are highly similar in terms of efficacy, safety and immunogenicity. The switch from Humira to BI 695501 had no impact on efficacy, safety and immunogenicity. These data, together with the analytical and the phase I data, suggest that BI 695501 and Humira are biosimilar and thus therapeutically equivalent.
Acknowledgments
The authors thank Liam Sebag-Montefiore and Tamara Bailey of Ashfield Healthcare Communications, part of UDG Healthcare, for medical writing support, which was funded by Boehringer Ingelheim.
Footnotes
Handling editor Tore K Kvien
Contributors SBC and DA were involved in the acquisition, analysis or interpretation of the data. All authors were involved in the critical revision of the manuscript, approval to submit and in agreement to be accountable.
Funding Boehringer Ingelheim provided the funding and was responsible for the conduct of this study.
Competing interests SBC and ECL received funding from Boehringer Ingelheim, study sponsor, as principal investigators of this study. AA-R and PAK have no competing interests to declare. NP, IS and DA are (or were) employees of Boehringer Ingelheim, study sponsor.
Patient consent Obtained.
Ethics approval The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the institutional review boards of all participating centres. All patients provided written informed consent.
Provenance and peer review Not commissioned; externally peer reviewed.