Article Text

Abstract
Objectives Adult-onset Still’s disease (AOSD) is a rare systemic autoinflammatory disease; its management is largely empirical. This is the first clinical study to determine if interleukin (IL)-18 inhibition, using the recombinant human IL-18 binding protein, tadekinig alfa, is a therapeutic option in AOSD.
Methods In this phase II, open-label study, patients were ≥18 years with active AOSD plus fever or C reactive protein (CRP) levels ≥10 mg/L despite treatment with prednisone and/or conventional synthetic disease-modifying antirheumatic drugs (DMARDs). Previous biological DMARD treatment was permitted. Patients received tadekinig alfa 80 mg or 160 mg subcutaneously three times per week for 12 weeks; those receiving 80 mg not achieving early predicted response criteria (reduction of ≥50% CRP values from baseline and fever resolution) were up-titrated to 160 mg for a further 12 weeks. The primary endpoint was the occurrence of adverse events (AEs) throughout the study.
Results Ten patients were assigned to receive 80 mg tadekinig alfa and 13 patients to the 160 mg dose. One hundred and fifty-five treatment-emerging AEs were recorded, and 47 were considered related to the study drug. Most AEs were mild and resolved after drug discontinuation. Three serious AEs occurred, one possibly related to treatment (toxic optic neuropathy). At week 3, 5 of 10 patients receiving 80 mg and 6 of 12 patients receiving 160 mg achieved the predefined response criteria.
Conclusions Our results indicate that tadekinig alfa appears to have a favourable safety profile and is associated with early signs of efficacy in patients with AOSD.
Trial registration number NCT02398435.
- adult onset still’s disease
- inflammation
- juvenile idiopathic arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Adult-onset Still’s disease (AOSD) is a rare non-familial, non-monogenic systemic inflammatory disease, the aetiology and pathogenesis of which remain unknown.1 2 AOSD belongs to the group of autoinflammatory disorders characterised by excessive innate immune responses. AOSD shares many similarities with systemic-onset juvenile idiopathic arthritis (SoJIA), but is approximately 10 times less frequent than its juvenile counterpart.1 3 The course of AOSD is heterogeneous with patients experiencing a monocyclic phase with complete resolution, and others with persisting or recurrent bouts of arthritis and systemic inflammation.4 The management of AOSD is largely empirical and includes the use of non-steroidal anti-inflammatory drugs (NSAIDs), systemic glucocorticoids and conventional synthetic (cs) disease-modifying anti-rheumatic drugs (DMARDs), such as methotrexate (MTX).5 Randomised clinical trials in SoJIA have demonstrated the efficacy of anticytokine therapies, including interleukin (IL)-1 and IL-6 antagonists.6 7 Similar strategies are used in AOSD, although the data are more scarce, including mainly retrospective studies,8–11 and only one randomised open clinical trial.12 The IL-1 antagonist, canakinumab, is indicated for the treatment of AOSD in patients who have responded inadequately to previous therapy with NSAIDs and systemic corticosteroids13; however, results from controlled clinical studies are not currently available.
IL-18 has been considered to play a major role among the inflammatory agents involved in AOSD pathogenesis.14 IL-18 is a proinflammatory cytokine of the IL-1 family that is produced by various cell types, including monocytes/macrophages.15 The biological activity of IL-18 is tightly controlled by IL-18 binding protein (IL-18BP), a naturally occurring inhibitor that binds IL-18 with high affinity.16 In AOSD, circulating levels of IL-18 were associated with clinical status and laboratory markers of disease activity.17 18 However, currently used immunoassays do not distinguish IL-18 complexed with IL-18BP (inactive) and unbound free IL-18 (active). Recently, by using a novel immunoassay that selectively measured biologically active IL-18, we showed that serum levels of free IL-18 were elevated in AOSD and correlated with clinical and biological markers of disease activity.19 The aim of the current study was to determine the safety and efficacy of blocking IL-18 with the administration of recombinant human IL-18BP (tadekinig alfa) in the treatment of AOSD. This clinical trial was the first to determine if IL-18 inhibition is a therapeutic option in AOSD.
Methods
Study participants
The study (ClinicalTrials.gov, number NCT02398435) was conducted between March 2015 and July 2016. Eligible patients were age 18 or older at baseline with AOSD according to the Yamaguchi criteria.20 Patients had active disease at baseline as defined by the presence of at least two Yamaguchi criteria at the screening visit plus either fever or elevated serum levels of C reactive protein (CRP ≥10 mg/L) despite being treated with prednisone at ≥5 mg daily for more than 1 month and/or csDMARDs (MTX at a dose of 10 mg per week for ≥3 months). Previous treatment with synthetic or biological DMARDs (bDMARDs) was allowed. All bDMARDs had to be discontinued before baseline, respecting specific washout periods (described in the online supplementary materials). Concomitant use of NSAIDs, prednisone and csDMARDs was allowed during the study. Specifically, the prednisone daily dosage could be maintained or tapered, but any increase was considered as treatment failure. Other inclusion and exclusion criteria are described in the online supplementary materials.
Supplementary file 1
All patients provided written informed consent before study participation. The protocol, informed consent and any accompanying material were approved by the ethics committees or institutional review board at each centre before study initiation.
Study design
This international, multicentre, open-label, dose-escalating phase II study included patients from 20 centres in Switzerland, France and Germany. Patients were subdivided into two groups that were sequentially treated with subcutaneous injections of either 80 mg or 160 mg tadekinig alfa three times per week for 12 weeks. Tadekinig alfa was available at a concentration of 80 mg/mL. Two injections of 80 mg tadekinig alfa were given to patients receiving tadekinig alfa at a dose of 160 mg. All injections were administered by trained study nurses throughout the study period. After the first five patients were assigned to the 80 mg group, and after the independent Data Safety Monitoring Board (DSMB) approval, patients were randomly assigned 1:1 to receive either 80 mg or 160 mg tadekinig alfa. Allocation to receive 80 mg was terminated after the 10th patient was enrolled into this group. The decision to continue with the study drug and to up-titrate to a higher dose was at the discretion of the DSMB. Early predicted criteria of response at 3 weeks were normalisation of body temperature and decrease by 50% of the baseline CRP levels or normalisation of CRP values to <5 mg/L. Response to therapy at 12 weeks was predefined as an improvement of joint count (both Swollen Joint Count (SJC) and Tender Joint Count (TJC) according to a 44-joint assessment) by ≥20% from baseline values, and a 70% decrease of CRP levels compared with baseline values (or reduction to normal levels) or normalisation of ferritin.
After 3 weeks, patients receiving tadekinig alfa 80 mg who did not achieve early predicted criteria of response were up-titrated to the 160 mg dose for a further 12 weeks of treatment. Dose increases to 160 mg for the 80 mg group, or to 320 mg for the 160 mg group, were also allowed after 3 weeks. A third group, 320 mg, was planned to be included within the study; however, it was decided not to enrol patients into this group since early efficacy was achieved in the 80 mg and 160 mg groups. Any dose increases from 80 mg to 160 mg or 160 mg to 320 mg were made at the treating physician’s discretion. Enrolled patients continued treatment through week 16, with a 4-week safety follow-up.
Assessments
Patients had regular scheduled visits at baseline (first tadekinig alfa administration) and weeks 1, 3, 6, 12 and 16.
The primary endpoint was the occurrence of adverse events (AEs) that were recorded throughout the study. The incidence, nature and severity of AEs, and abnormal laboratory tests were reported.
Secondary endpoints comprised clinical and biological signs of efficacy, including the evolution of body temperature, skin rash, number of SJC and TJC, patient global assessment, physician global assessment, pain assessment on a visual analogue scale (VAS), fatigue assessment on the VAS and dosage of prednisone with respect to baseline. Laboratory assessments included serum levels of CRP, serum amyloid A (SAA), ferritin, free IL-18, IL-6, tumour necrosis factor, IL-1 receptor antagonist (IL-1Ra), S100A8/9, S100A12 and blood leucocyte and granulocyte counts. All laboratory tests were performed in a centralised laboratory (MLM Medical Labs GmbH, Moenchengladbach, Germany; see online supplementary materials).
A small pharmacokinetic study was also performed (see online supplementary materials).
Statistical analysis
For AEs, the statistical analyses were descriptive and the reports included standard summary tables (including mean, SD, median, minimum, maximum or counts/percentages).
For secondary endpoints, to better assess efficacy, we used per-protocol analyses by imputing missing values only from week 6 to week 12 (last observation carried forward). If patients stopped earlier, they were excluded from the analyses. Continuous variables were compared over time using Wilcoxon signed-rank test and across dosages using Wilcoxon rank-sum test. Categorical variables were compared over time using exact McNemar’s test and across dosages using Fisher’s exact test. All analyses and tabulations were performed using R V.3.4.1.
Results
Twenty-three of the 32 screened patients were included in the study. Ten and 13 patients were assigned to receive doses of either 80 mg (group 1) or 160 mg (group 2) tadekinig alfa three times per week, respectively (figure 1). The baseline characteristics of the patients included in the two groups are described in table 1. In the overall study population of 23 patients, arthritis and arthralgia (SJC and TJC) were present in 19 patients at baseline, neutrophilia in 14 patients, skin rash in 13 patients and fever in only 2 patients. Six patients had an early disease onset within 6 months prior to study inclusion, whereas all others had either chronic or polycyclic courses. Twenty-two patients had previously received glucocorticoids up to a median dose (IQR of 30 mg daily, 8.5–47.5). Thirteen patients had at least one course of treatment with a csDMARD and nine patients with at least one biological agent. One patient had received no prior glucocorticoids or csDMARDs. During the study, 21 patients received concomitant glucocorticoids at a median (IQR) baseline prednisone dosage of 15 mg daily (7.5–20), and 9 patients received a concomitant csDMARD, including 7 patients treated with MTX at a median (IQR) dosage of 17.5 mg weekly (15–22.5). By chance, patients recruited in group 2 were younger (35 vs 49.5 years) with shorter disease duration (11.6 vs 25.5 months) and lower prednisone daily dose (15 vs 35 mg) than in group 1.
Demographic and baseline characteristics of patients with AOSD treated with tadekinig alfa

Patient disposition. *One responder and five non-responders. †One patient (responder) was up-titrated to 320 mg. AOSD, adult-onset Still’s disease; ISR, injection site reaction; SAE, serious adverse event.
One patient from group 2 discontinued after 1 week of therapy due to an injection site reaction (ISR) and was excluded from the efficacy analysis.
Safety analyses
The description of AEs by System Organ Class is shown in table 2. One hundred and fifty-five treatment-emergent AEs were recorded; 47 were considered related to the study drug by the treating physicians. ISRs, upper airway infections and arthralgia were the most common AEs. Three and 10 patients showed manifestations of local ISRs in groups 1 and 2, respectively. Some patients had more than one episode and most were considered mild. Two patients in group 2 had moderately severe ISR.
Most frequent AEs, >5% of the patients, by SOC and preferred term
Three serious AEs (SAEs) occurred during the study; two were considered, by the treating physicians, as not related to tadekinig alfa (one episode of gastroenteritis and one severe back pain due to spondylolisthesis) and one as possibly related (toxic optic neuropathy). Case details of the toxic optic neuropathy SAE are provided within the online supplementary materials.
AEs led to the permanent discontinuation of the study in four patients, including three cases of ISR, one with 80 mg and two with 160 mg tadekinig alfa doses, respectively, and one case of SAE due to toxic optic neuropathy (details within the online supplementary materials). The patient who was up-titrated from 160 mg to 320 mg tadekinig alfa experienced no AE besides mild ISR.
Efficacy analyses
Among the patients available for clinical evaluation, five patients treated with the 80 mg dose (group 1), and six patients treated with the 160 mg dose (group 2) achieved the predefined response criteria, including a reduction of ≥50% CRP values from baseline and resolution of fever (table 3). As per the study protocol, five patients initially treated with 80 mg tadekinig alfa who did not achieve the response criteria at week 3 were up-titrated to receive 160 mg tadekinig alfa for an additional 12 weeks. Two responders, according to predefined study criteria at week 3, were also up-titrated: one patient from 80 mg to 160 mg and one patient from 160 mg to 320 mg tadekinig alfa, at the discretion of the treating physician. Since at least four patients in the 160 mg group achieved early predicted criteria of response, no patients were assigned to receive the higher dose based on non-response.
Clinical and biological signs of response
In patients without arthritis at baseline, clinical response was solely based on the results of laboratory tests. The 12-week efficacy analysis was first carried out including all patients with data at 12 weeks of tadekinig alfa treatment (table 3). Of the four patients treated with tadekinig alfa 80 mg throughout the study, two achieved the response criteria. Of 12 patients initially treated with 160 mg tadekinig alfa, 7 patients achieved the response criteria. Eight of the 18 patients who received 160 mg tadekinig alfa, either from the study start or after 3 weeks of treatment with 80 mg tadekinig alfa, achieved the response criteria at week 12 (table 3). Both patients who up-titrated to 160 mg or 320 mg continued to be responders after 12 weeks. None of the non-responding patients from group 1 achieved the response criteria after 12 weeks of therapy with tadekinig alfa 160 mg. Of note, the response rate in patients with systemic manifestations but without synovitis (seven patients) did not differ from the whole group. The patient without prior glucocorticoid or DMARD treatment responded to tadekinig alfa (see online supplementary materials).
Additional secondary endpoints
Skin rash showed improvement over time versus baseline (figure 2), with 13/23 patients having skin rash at baseline, and only 6 patients at 12 weeks (P=0.02). Patient-reported and physician-reported outcomes also showed a general trend towards improvement (online supplementary table S1).
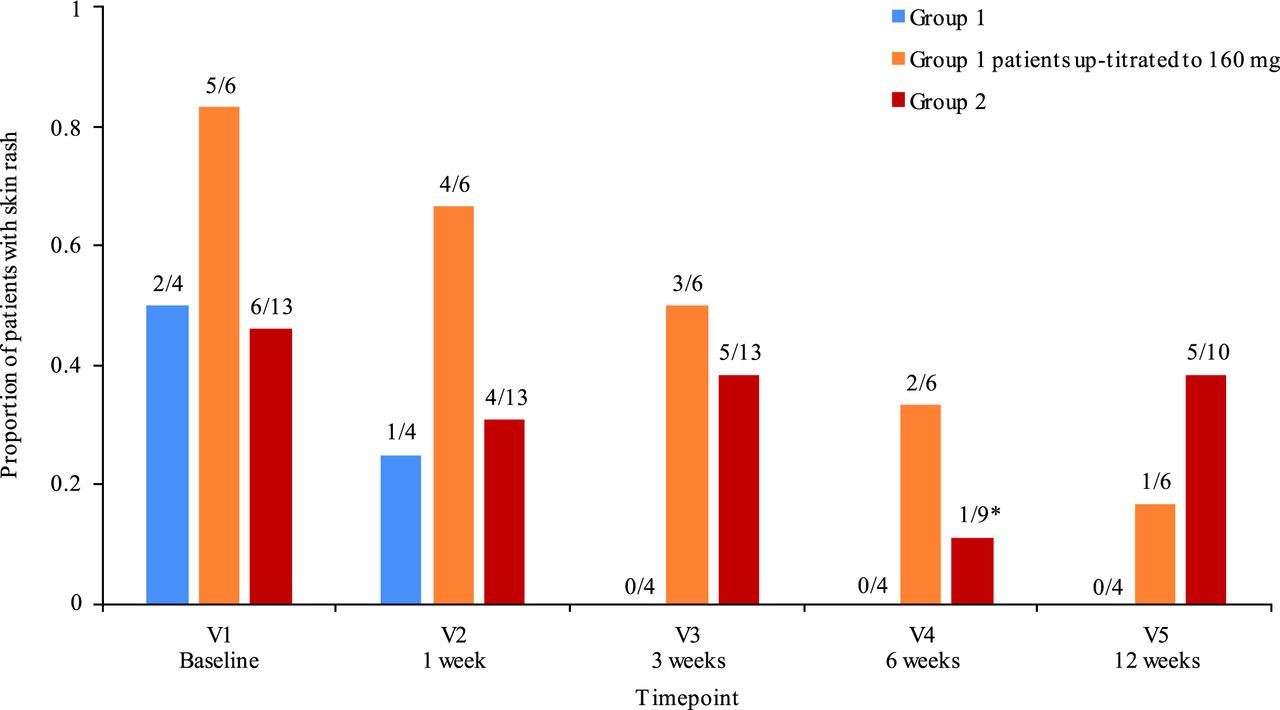
Evolution of skin rash over time. Presented fractions show number of patients with rash at each V/total number of patients. *Four patients did not have V4, but had V5 or V6 (early termination V). V, visit.
Overall, median prednisone dosage was decreased from 12.5 mg/day at baseline to 10 mg/day at 12 weeks (P=0.01). The decrease in prednisone was larger among the responders at 12 weeks with a tapering of 12.8 mg, compared with only 1 mg in the non-responders, although this difference was not statistically significant (P=0.23).
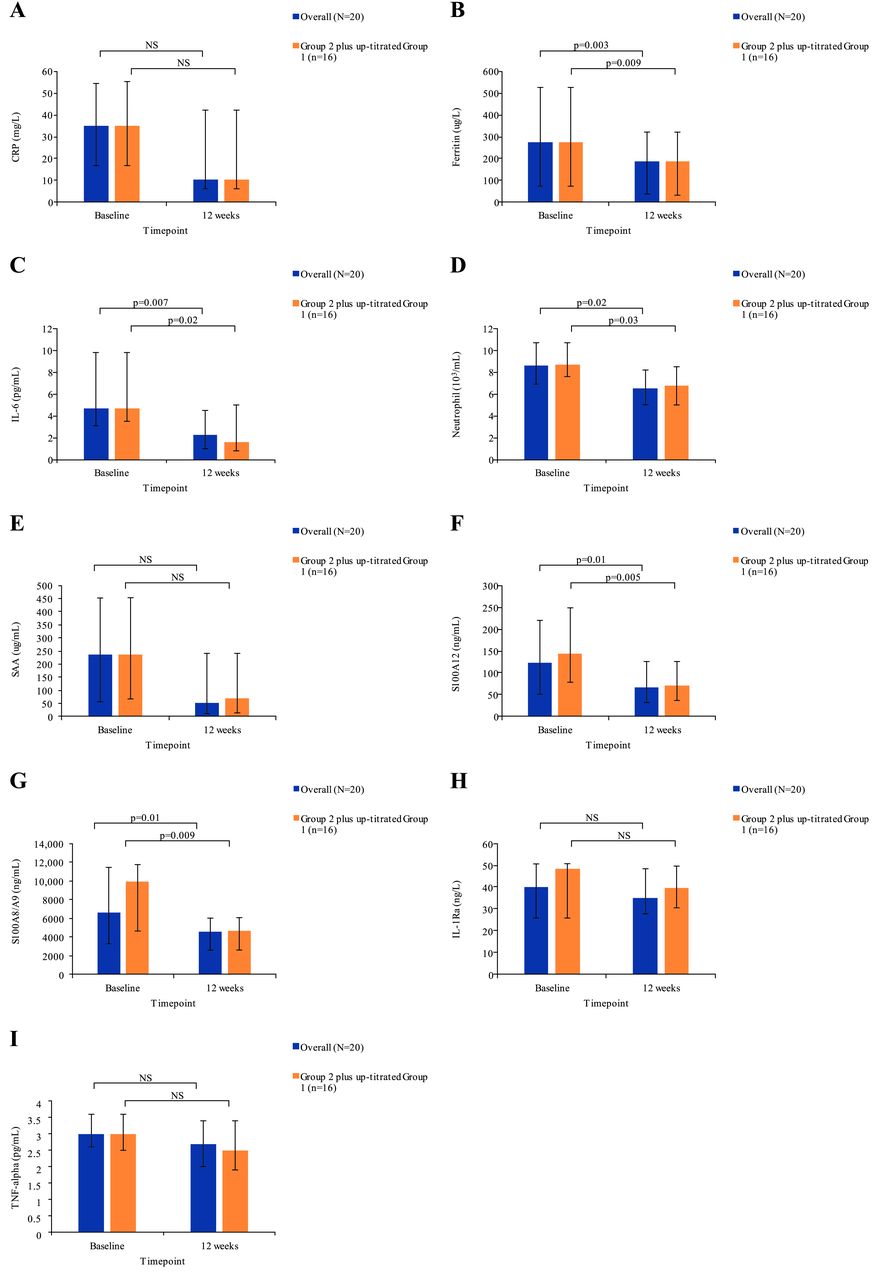
Biomarker levels decreased at week 12 compared with baseline levels (figure 3A–I; online supplementary table S2). Serum levels of free IL-18 were detected in seven patients at baseline. Among these patients, four exhibited a clinical response. Free IL-18 was undetectable in all these patients at the final blood assessment, whereas free IL-18 remained elevated in two of three patients who failed to respond to tadekinig alfa. In the overall population, as well as in patients from group 2 and in patients from group 1 up-titrated to the 160 mg dosage, the levels of ferritin, IL-6, neutrophils, S100A8/9 and S100A12 significantly decreased at week 12, as compared with baseline levels. Levels of CRP, SAA, ferritin, IL-6, S100A8/9 and S100A12 were significantly decreased in patients from group 2 (figure 3; online supplementary table S2). Elevated transaminase levels were present at baseline in two patients (>3-fold upper normal limits) and normalised with tadekinig alfa therapy in both patients.

{kind=link}
{kind=link}
{kind=link}
Evolution of serum biomarker levels by group and time point: (A) CRP; (B) ferritin; (C) IL-6; (D) neutrophil; (E) SAA; (F) S100A12; (G) S100A8/A9; (H) IL-1Ra; (I) TNF-alpha. Data are median (IQR). The 20 patients included in the overall group comprised 17 patients who had baseline values and at week 12, and 3 patients with values at week 6, but who discontinued thereafter. Three patients who discontinued at earlier time points were not included. Group 2 plus up-titrated group 1, n=16. CRP, C reactive protein; IL, interleukin; Ra, receptor antagonist; NS, not significant; SAA, serum amyloid A; TNF, tumour necrosis factor.
Results of the pharmacokinetic study are reported in the online supplementary materials.
Discussion
This phase II clinical trial examined for the first time the safety and efficacy of IL-18 blockade in patients with AOSD. Most patients had previously used glucocorticoids; approximately half had received csDMARDs and one-third bDMARDs. The results show that tadekinig alfa at doses of 80 mg and 160 mg three times per week appeared to have a favourable safety profile. In addition, we observed early signs of clinical and laboratory marker efficacy with response rates of 50%, irrespective of the tadekinig alfa dosage in this heterogeneous population of difficult-to-treat patients.
Most of the AEs were mild and resolved after drug discontinuation. The 60-year-old patient in which toxic optic neuropathy occurred suffered from hypertension, pulmonary emphysema and had serious thrombotic episodes prior to study participation. This AE was unexpected according to results of previous clinical studies. The DSMB reviewed the case and questioned the physician’s conclusion that it was possibly drug related, suggesting that other, more likely, explanations had not been ruled out. The DSMB considered that there was insufficient information to draw any firm conclusion since there had been insufficient exploration to discard the diagnosis of ophthalmic vein thrombosis in this patient. ISRs were more frequently observed in patients receiving tadekinig alfa 160 mg.
The safety and efficacy of tadekinig alfa has been examined in two double-blinded, placebo-controlled, phase Ib clinical trials in 36 patients with rheumatoid arthritis and 35 patients with psoriasis (unpublished results). The safety profile showed that the most commonly reported AEs were ISRs that were mild in most cases. Local tolerability tended to worsen with increasing doses. Tadekinig alfa was administered subcutaneously at doses ranging from 80 mg to 350 mg three times per week based on the 30-hour half-life of tadekinig alfa for 6 weeks. These studies did not show any definite signs of efficacy.
In the current study, the results at week 3 did not show any difference in response between 80 mg and 160 mg tadekinig alfa doses. Furthermore, all non-responders who were up-titrated from 80 mg to 160 mg did not reach a subsequent clinical response. These results suggest that the tadekinig alfa 80 mg dose already has a meaningful clinical effect.
One patient was included despite not meeting the inclusion criteria of having been previously exposed to glucocorticoids, NSAIDS and/or csDMARDs since this case provides additional information on safety, our primary endpoint. This patient responded to tadekinig alfa without the addition of glucocorticoids or csDMARDs. Most importantly, following the discontinuation of tadekinig alfa at week 12, the patient experienced a disease flare.
Consistent with the positive effect of IL-1 or IL-6 targeting in SoJIA therapies, inhibition of these pathways has been studied in AOSD. Tocilizumab, a humanised monoclonal antibody against IL-6 receptor alpha, significantly reduced articular and systemic manifestations, acute-phase markers and prednisone dosage in patients with AOSD.8 In 140 patients with AOSD treated with anakinra, a human recombinant IL-1Ra, systemic and articular manifestations improved in most patients, and a glucocorticoid-sparing effect and a significant reduction in the number of patients on csDMARDs were observed.10 Our results show that IL-18 inhibition offers another possibility of therapy within the scope of anticytokine treatment for the management of AOSD.
The fact that only two patients had fever at baseline was unexpected. However, all patients had fever at some point during the disease course. It is plausible that the inclusion of some patients with long-standing disease may explain this observation. Some disease manifestations may also have been partly controlled by previous therapies. However, to avoid a carryover effect of former bDMARDs, a long washout period (ie, 6 months for canakinumab) was required prior to study inclusion.
Our study is the first prospective study examining a drug with a completely new mode of action in AOSD. Another strength of our study is the inclusion of patients with ‘difficult to treat’ disease, including a large percentage of patients previously treated with bDMARDs. Furthermore, several patients had various comorbidities. Thus, our findings provide important results regarding the safety of IL-18 targeting with tadekinig alfa in AOSD. An open-label design, absence of a control group and heterogeneity within the study population are important limitations in the assessment of a treatment’s efficacy. However, the clinical response in a group of patients with chronic disease despite the use of DMARDs and inclusion of objective measures, such as biomarkers of inflammatory responses, provide supportive data for treatment efficacy.
In conclusion, our results show that tadekinig alfa appears to have a favourable safety profile and is associated with early signs of efficacy in AOSD, thus warranting further clinical investigation.
Acknowledgments
We are grateful to the patients and investigators who participated in the phase IIb study for their involvement and support. We also thank the members of the AB2 Bio Ltd tadekinig alfa team (in particular, Greg Del Val, Lalla Myriam Mercier-Lamrani, Jon Lacy and Fernando Cunha) for their input. We thank the members of the Data and Safety Monitoring Board—Ernest Choy, MD, PhD (Cardiff, UK), Michele Bombardieri, MD, PhD (London, UK) and Roberta Priori, MD, PhD (Rome, Italy)—for their invaluable guidance. Editorial support was provided by Sally Gray (Medical Writer) of ELM Medical, Stafford, UK; this support was funded by AB2 Bio Ltd.
References
Footnotes
Handling editor Josef S Smolen
Contributors CG, BF, JR, JM, AS and EJS contributed to the conception and design of this study. CG, BF, JR, FS, EF, IK, EH, JM, TS, MAH, TM, BH, PL and HSK recruited patients into the study and participated in data collection. CG, FS, EH, DSC, AS and EJS contributed to the data analysis. All authors contributed to data interpretation, critically reviewed the article for important intellectual content and approved the final draft for submission.
Funding This study was sponsored by AB2 Bio Ltd, EPFL Innovation Park, Lausanne, Switzerland.
Competing interests CG has received grants and personal fees from AB2 Bio Ltd, grants and personal fees from Roche and Pfizer, and personal fees from Merck Sharp & Dohme (MSD), Novartis, Sanofi and AbbVie; BF has received grants from AbbVie, MSD, Pfizer and Roche, and personal fees from AbbVie, Biogen, Bristol-Myers Squibb (BMS), Celgene, Janssen, Lilly, MSD, MEDAC, Nordic, Pfizer, Roche, Swedish Orphan Biovitrum AB (publ) (Sobi), Novartis and Union Chimique Belge (UCB); FS has received grants from AB2 Bio Ltd; EF has received financial and non-financial support from AB2 Bio Ltd; IK has received personal fees from AbbVie, Actelion, BMS, Celgene, GlaxoSmithKline, Janssen, Novartis, Pfizer, Roche and Sobi; TS has received grants and personal fees from Pfizer and Roche, and personal fees from AbbVie, Biogen, BMS, Lilly, MSD, Novartis, Sanofi and UCB; BH has received personal fees from AbbVie, BMS, Novartis, Roche, Pfizer, Celgene and MSD; PL has received non-financial support from AB2 Bio Ltd; DSC has received grants from AB2 Bio Ltd and personal fees from Pfizer, BMS and Janssen; AS and EJS are employees of AB2 Bio Ltd.
Ethics approval The study was approved by each centre’s institutional review board or ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Further data on SF12 can be shared upon request to the sponsor of the study.