Article Text

Abstract
Objectives Several common and rare risk variants have been reported for systemic sclerosis (SSc), but the effector cell(s) mediating the function of these genetic variants remains to be elucidated. While innate immune cells have been proposed as the critical targets to interfere with the disease process underlying SSc, no studies have comprehensively established their effector role. Here we investigated the contribution of monocyte-derived macrophages (MDMs) in mediating genetic susceptibility to SSc.
Methods We carried out RNA sequencing and genome-wide genotyping in MDMs from 57 patients with SSc and 15 controls. Our differential expression and expression quantitative trait locus (eQTL) analysis in SSc was further integrated with epigenetic, expression and eQTL data from skin, monocytes, neutrophils and lymphocytes.
Results We identified 602 genes upregulated and downregulated in SSc macrophages that were significantly enriched for genes previously implicated in SSc susceptibility (P=5×10−4), and 270 cis-regulated genes in MDMs. Among these, GSDMA was reported to carry an SSc risk variant (rs3894194) regulating expression of neighbouring genes in blood. We show that GSDMA is upregulated in SSc MDMs (P=8.4×10−4) but not in the skin, and is a significant eQTL in SSc macrophages and lipopolysaccharide/interferon gamma (IFNγ)-stimulated monocytes. Furthermore, we identify an SSc macrophage transcriptome signature characterised by upregulation of glycolysis, hypoxia and mTOR signalling and a downregulation of IFNγ response pathways.
Conclusions Our data further establish the link between macrophages and SSc, and suggest that the contribution of the rs3894194 risk variant to SSc susceptibility can be mediated by GSDMA expression in macrophages.
- systemic sclerosis
- macrophage
- GSDMA
- eQTL analysis
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Systemic sclerosis (SSc) is an intractable chronic autoimmune disease of unknown aetiology with high clinical heterogeneity and mortality rates. SSc is characterised by complex inflammatory, vascular and fibrogenic interactions occurring in multiple systems and tissues.1 Among the cellular populations contributing to the pathogenesis of SSc, monocytes/macrophages have been suggested to play a key role in initiating and/or perpetuating the disease,2 but their specific role and importance are still unclear. Candidate gene and genetic screen studies have begun to elucidate the genetic architecture of SSc3; for instance, genome-wide association studies (GWAS) and whole exome sequencing (WES)4 have reported numerous genes associated with susceptibility to SSc or to SSc subphenotypes and related traits.5 However the functional and cellular context of many genes and variants associated with SSc remains poorly understood. In macrophages, gene sets representative of macrophage activation have been used for enrichment analyses in expression profiles obtained from SSc-associated tissues,6 but the direct link between SSc disease variants and macrophage transcriptome remains to be elucidated.
Here we integrate differential expression and expression quantitative trait locus (eQTL) analyses in monocyte-derived macrophages (MDMs) from patients with SSc and healthy controls, revealing (1) changes in macrophage transcriptome as an important contributor to SSc and (2) upregulation and cis-regulation of GSDMA (a candidate gene for SSc susceptibility) contributing to disease risk in macrophages but not in skin.
Methods
Sample collection and clinical details
Patients with SSc met the American Rheumatism Association preliminary criteria for a diagnosis of SSc.7 The study was carried out with a total of 57 patients who attended the rheumatology clinic at the Royal Free Hospital exhibiting SSc with subgroups of limited cutaneous SSc (lcSSc) and diffuse cutaneous SSc (dcSSc). Patients with overlap features of another autoimmune rheumatic disease were excluded. Cases were classified as lcSSc or dcSSc according to extent of skin thickening8 and reflected the expected serological and clinical characteristics of the cohort that have been detailed in previous publications.9 Patients were receiving standard treatments for SSc in line with current European League Against Rheumatism recommendations.10 Thus, 11 patients were receiving low-dose prednisolone, 9 had received methotrexate, 18 received mycophenolate and 2 had received other potential disease-modifying agents (cyclophosphamide and rituximab, respectively). As expected, immunosuppression was more frequently used in cases with diffuse skin disease. Blood samples were also collected from 15 healthy control subjects. Details for all the samples included in this study can be found in online supplementary table S1. All subjects gave written informed consent. Blood (25 mL) was drawn from all patient and control samples using standardised phlebotomy procedures into sodium citrate tubes.
Supplementary file 2
Isolation of MDMs
Human MDMs were differentiated from total blood from patients with SSc and healthy donors using gradient separation (Histopaque 1077, Sigma) and adhesion purification. Following Histopaque separation, peripheral blood mononuclear cells were resuspended in RPMI (Life Technologies), and monocytes were purified by adherence for 1 hour at 37°C, 5% CO2. The monolayer was washed three times with Hank’s Balance Salt Solution (HBSS) to remove non-adherent cells, and monocytes were matured for 5 days in RPMI containing 100 ng/mLmacrophage colony-stimulating factor (M-CSF) (PeproTech, London, UK) and 10% fetal calf serum (Labtech International). Macrophage purity was confirmed by immunohistochemical assessment of CD68 and >99% cells were CD68+.
RNA extraction and RNA sequencing
Total RNA was extracted from human monocyte-derived macrophages (hMDMs) using TRIzol (Invitrogen) and RNeasy Mini Kit (Qiagen) according to manufacturers’ instructions, with an additional purification step by on-column DNase treatment using the RNase-Free DNase Kit (Qiagen) to ensure elimination of any genomic DNA. The integrity and quantity of total RNA were determined using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific) and Agilent 2100 Bioanalyzer (Agilent Technologies). In total 500 ng of total RNA was used to generate RNA-sequencing (RNA-seq) libraries using TruSeq RNA Sample Preparation Kit (Illumina) according to the manufacturer’s instructions. Briefly, RNA was purified and fragmented using poly-T oligo-attached magnetic beads using two rounds of purification followed by the first and second complementary DNA (cDNA) strand synthesis. Next, cDNA 3' ends were adenylated and adapters ligated followed by 15 cycles of library amplification. Finally, the libraries were size-selected using AMPure XP Beads (Beckman Coulter), purified and their quality was checked using Agilent 2100 Bioanalyzer. Samples were randomised to avoid batch effects, and multiplexed libraries were run on a single lane (six samples/lane) of the HiSeq 2500 platform (Illumina) to generate 100 bp paired-end reads. An average coverage of 64M reads per sample was achieved. The RNA-seq data have been deposited in NCBI’s Gene Expression Omnibus (GEO) database (GEO Series accession number GSE104174).
Quantitative reverse transcription PCR analysis
cDNA was obtained from 500 ng of total RNA using the Bio-Rad iScript Kit (Bio-Rad, Hertfordshire, UK) according to the manufacturer’s instructions. Quantitative reverse transcription PCR reactions were performed using the ViiA 7 Real-Time PCR System (Life Technologies). A total of 10 ng of cDNA per sample was used for PCR using Brilliant II SYBR Green qPCR Master Mix (Agilent Technologies). QuantStudio Real Time PCR Software (Life Technologies) was used for the determination of treshold cycle (Ct) values. Results were analysed using the comparative Ct method,11 and each sample was normalised to the reference gene (HPRT) to account for any cDNA loading differences. Results are expressed as mean±SEM, and statistical analysis was performed using Student’s t-test.
Genotyping
DNA was isolated from 1 mL of whole blood of 71 samples (57 patients with SSc and 14 controls) using Gentra Puregene Blood Kit (Qiagen). Genotyping was performed on the Illumina Infinium Omni2.5–8 1.3 platform, which resulted in 2 372 784 genotype calls (Illumina GenomeStudio V.1.9.4 software).
RNA-seq and genotype data processing and detailed description of all the analyses included in this work can be found in the online supplementary file.
Supplementary file 1
Results
Differential expression analysis of SSc and control MDMs
Differential expression analysis of MDMs expression profiles, in patients with SSc (n=57) and controls (n=15) (online supplementary figure S1), identified 170 upregulated and 432 downregulated genes in SSc, respectively (a total number of 602 genes, false discovery rate (FDR) <0.1; figure 1A and online supplementary table S2). Quantitative PCR analysis validated the changes detected by RNA-seq for a subgroup of genes (online supplementary figure S2). We revealed hundreds of genes associated with SSc in MDMs, including genes previously implicated in the genetic aetiology of the disease (figure 1B). In addition, 145 (25%) out of these 602 differentially expressed (DE) genes have been reported to interact functionally at the protein and pathway levels (figure 1C). Consistent with previously proposed biological processes and pathways associated with SSc,3 the upregulated genes showed significant enrichment for unfolded protein response, epithelial mesenchymal transition and DNA repair, whereas the downregulated genes showed enrichment for innate immune response-related processes (figure 1D), such as interferon response and allograft rejection, including genes previously linked to SSc (eg, IL2RB,12 TNFAIP3, HLA-DQA1, HLA-DRB1 3). Consistent with the previous SSc transcriptomics analysis in skin13 and in fibroblasts from patients with SSc-associated interstitial lung disease,14 the majority of the DE genes were downregulated in MDMs. The SSc macrophage transcriptome showed enrichment for genes involved in increased metabolic rates (glycolysis, hypoxia and mammalian target of rapamycin (mTOR) signalling), which have been previously linked with a proinflammatory activation profile.15
Supplementary file 3
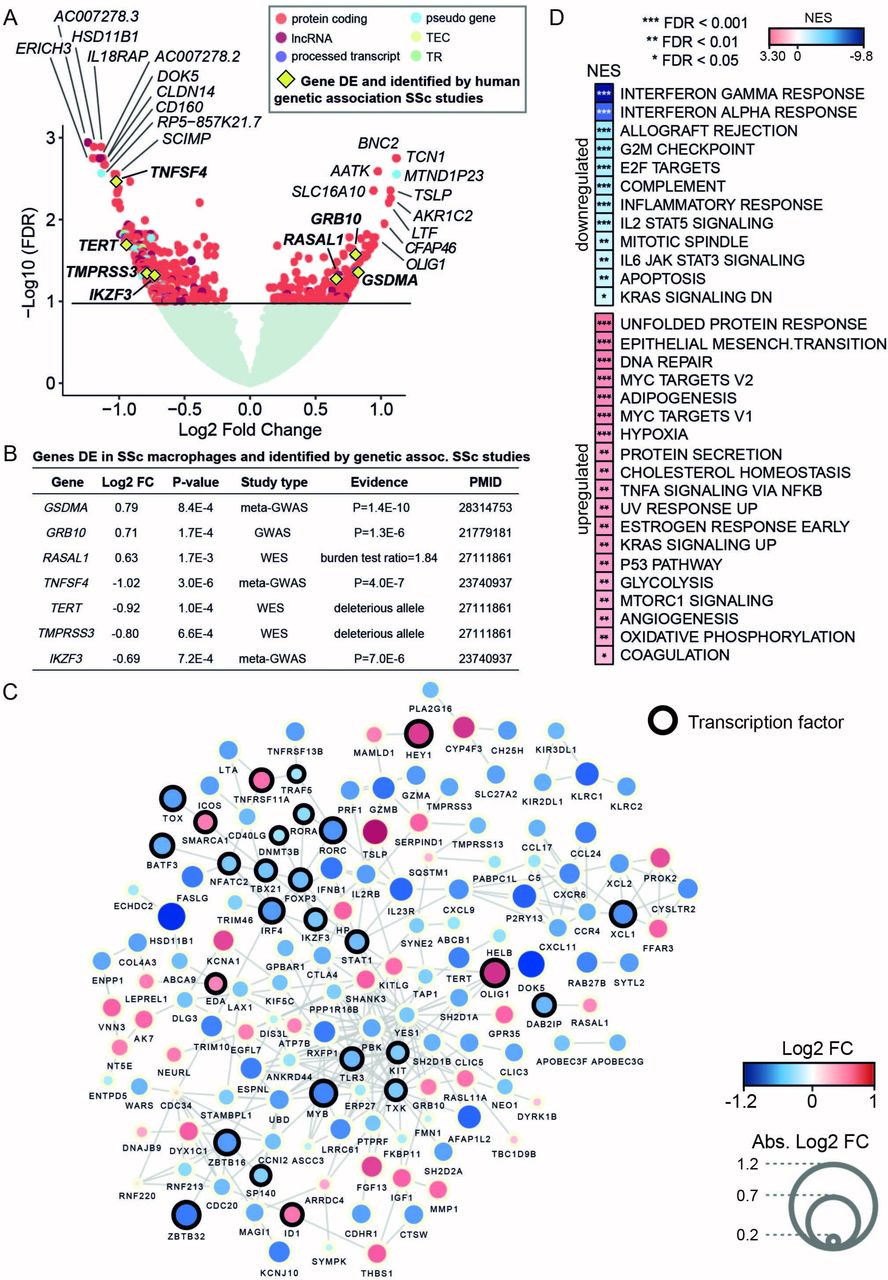
RNA sequencing differential expression analysis between monocyte-derived macrophages (MDMs) from patients with systemic sclerosis (SSc) and healthy controls provides evidence for the involvement of macrophages in SSc and related cellular processes. (A) Volcano plot with differential expression results. Gene names of the top 10 upregulated and downregulated genes are included. Genes previously identified in SSc human genetic association studies are also highlighted (yellow diamond). Genes with no significant differential expression are displayed in light green, whereas differentially expressed (DE) genes (false discovery rate (FDR) <0.1) are displayed coloured by gene type (TEC denotes gene to be experimentally confirmed and TR denotes T cell receptor genes). (B) Summary of the DE genes in SSc MDMs that have been previously found to be associated with SSc susceptibility by human genetic studies. The table includes differential expression test statistics in MDMs (log2 fold change (FC) and P value) and information about the SSc genetic study in which the gene had previously been reported (ie, study type and the provided evidence for involvement with SSc susceptibility). (C) Network with known protein-protein and databases interactions (edges) between the DE genes (nodes) identified in SSc MDMs. Gene size and colour are mapped to log2 FC. Only the genes with reported connections are displayed here (see Methods). (D) Functional processes (from Hallmark database) enriched in the set of DE genes in SSc MDMs were computed by gene set enrichment analysis (GSEA).33 Normalised enrichment scores (NES) denote the upregulation and downregulation enrichment strength. FDR levels for the GSEA are also included. GWAS, genome-wide association studies; WES, whole exome sequencing.
To investigate whether the genes dysregulated in MDMs have been previously implicated with genetic susceptibility to SSc, we queried the National Human Genome Research Institute (NHGRI) GWAS Catalog5 and looked for genes identified by WES.4 We found that 10% of the genes previously associated with SSc overlapped with the set of DE genes detected in MDMs, representing a significant enrichment with respect to genome-wide expectation (10% observed overlap vs 2% expected overlap, Fisher’s exact test P=5×10−4; see online supplementary methods). Among the set of upregulated genes, we identified GSDMA and GRB10, which have previously been associated with SSc susceptibility16 and subphenotypes of SSc by GWAS,17 respectively. We also found RASAL1, a gene identified by WES that is enriched for deleterious variants in dcSSc.4 The set of downregulated genes included IKZF3 and TNFSF4 , both identified in a meta-GWAS SSc study,18 as well as TERT and TMPRSS3, candidate genes for dcSSc identified by WES.4 Therefore our DE analysis from patients with SSc and controls revealed hundreds of genes associated with SSc in MDMs, including genes previously implicated in the genetic aetiology of the disease.
Genetic regulation of macrophage gene expression in SSc
We carried out genome-wide cis-acting eQTL mapping in the cohort of patients with SSC (15 433 genes used as input; see online supplementary methods), which yielded 683 loci regulating the mRNA abundance of 270 genes in MDMs (genome-wide cis-eQTLs with FDR <5%; online supplementary table S2). The cis-regulated genes were nominally enriched for similar processes detected in the set of DE genes, such as interferon gamma (IFNγ) response and major histocompatibility complex class II protein complex (gene set enrichment analysis, P<0.02; online supplementary table S2). To identify cis-regulated genes associated with SSc, we integrated the eQTL data with the results of differential expression analysis (602 genes significantly differentially expressed with FDR <10%), which shortlisted five candidates, GSDMA, MMP1, AC004148.2, APOBEC3C and NMRK1, as the only genes that are both DE and cis-regulated in SSc MDMs (figure 2A). Among these candidates, GSDMA (Gasdermin A, a member of the Gsdm gene family that is required for tumour necrosis factor-α-induced apoptosis in mouse19) shows the highest upregulation in SSc MDMs. Furthermore, GSDMA is cis-regulated by the single nucleotide polymorphism (SNP) rs3859192 and is an established susceptibility gene for SSc (identified by the largest current transethnic meta-analysis comprising 4436 cases and 14 751 controls).16 Moreover, various genetic variants at the GSDMA locus have been previously associated with other autoimmune diseases with a proposed macrophage component, including asthma,20 rheumatoid arthritis,21 ulcerative colitis22 and Crohn’s disease.23 In contrast with the strong association of macrophage GSDMA expression with SSc reported here (P=8.4×10−4; figure 2A), GSDMA expression levels do not change in the skin of SSc (both diffuse and limited) in three independent studies (figure 2B). Despite GSDMA being most highly expressed in the skin as compared with 53 primary tissues/cell types analysed24 and the large sample size used for the cis-eQTL analysis (n≥250), the gene is not cis-regulated in the skin (normal and sun-exposed) (figure 2C).

{kind=link}
{kind=link}
Study of cis-regulated genes in systemic sclerosis (SSc) monocyte-derived macrophages (MDMs). (A) Manhattan plot with all the cis-eQTL results. Differentially expressed genes in SSc MDMs (false discovery rate (FDR) <0.1) that are also cis-regulated (FDR <0.05) are highlighted in orange (five genes). Expression levels of these genes in SSc and control MDMs are displayed in boxplots (P refers to the P value of the differential expression test; see online supplementary methods). Expression levels of these five genes in patients with SSc according to the genotype of the cis-regulatory SNP (x-axis, the number refers to the number of copies for the minor allele) are shown in boxplots (P refers to the P value of the cis-eQTL; see online supplementary methods). (B) GSDMA expression levels in the skin in three cohorts of patients with SSc and controls (first two boxplots are cohorts of diffuse patients with SSc, whereas the third boxplot refers to patients with limited SSc). GEO refers to Gene Expression Omnibus database followed by the database accession number for each skin data set. P refers to t-test P value (two-tailed). N refers to the samples size in each group. (C) GSDMA expression levels in MDMs (from this study, indicated with asterisk) alongside with expression from all tissues/cell types included in Genotype-Tissue Expression (GTEx) database24 (only tissues/cell types with GSDMA median transcripts per kilobase million (TPM) levels >0.5 are displayed). We include at the top of the graph the tissues/cell types in which rs3859192 has been shown to regulate GSDMA levels in the GTEx database (both effect size and cis-eQTL P value are shown). Among the tissues where GSDMA is significantly cis-regulated (grey background), GSDMA is most highly expressed in SSc macrophages (highlighted with yellow background). In the case of the macrophage data, eQTL refers to the results presented in panel (A). (D) Overview of the genomic region on chromosome 17 centred on the GSMDA gene where we report the SNPs associated with GSDMA expression levels in macrophages (top eQTL SNP rs3859192). y-axis (left), significance of the eQTL in SSc macrophages is reported using FDR. y-axis (right), recombination rate between the SNPs. SNPs are displayed coloured by linkage disequilibrium (LD). Both LD and recombination rate are estimated from 1000 genomes (March 2012) in the European population. The location of the risk variant for SSc previously reported by Terao et al 16 (rs3894194) is also indicated (this SNP is not genotyped in the cohort used in this MDMs study). (E) Summary of the regulatory information (methylation, acetylation and DNase hypersensitivity) included in the Roadmap Epigenomics and ENCODE projects27 for the cis-eQTL SNP for GSDMA (rs3859192) detected in SSc macrophages (see online supplementary file for details). Red box, monocyte and monocyte-derived cell types. eQTL, expression quantitative trait locus; GWAS, genome-wide association studies; HSMM, human skeletal muscle myoblasts; SNP, single nucleotide polymorphism.
To investigate whether the cis-regulation of GSDMA expression exists in a wider immune-cell context, we next assessed the genetic regulation of GSDMA expression in monocytes,25 neutrophils and lymphocytes.26 We did not find significant cis-regulation of GSDMA in basal (unstimulated) monocytes. Notably, the transcript was cis-regulated in monocytes stimulated with lipopolysaccharide at two time points (2 hours and 4 hours) and in IFNγ-stimulated (24 hours) monocytes25 (online supplementary table S4). These cis-regulatory SNPs found in stimulated monocytes included the cis-eQTL SNP rs3859192 found in MDMs from patients with SSc (figure 2D). No significant cis-regulation of GSDMA has been detected in neutrophils or lymphocytes.26 These results suggest that the functional relevance of GSDMA expression may be attributed to the monocyte/macrophage subset of the innate immune response.
Supplementary file 5
Our eQTL analysis revealed upregulation and significant cis-regulation of GSDMA mRNA levels in SSc MDMs (figure 2C), the latter is exerted by an intronic SNP rs3859192 (figure 2D). This eQTL SNP rs3859192 is in linkage disequilibrium (LD) with the risk variant rs3894194, which was found to be associated with SSc by transethnic meta-analysis.16 Specifically, the LD between rs3859192 and rs3894194 (estimated from 1000 genomes database) in European and African populations (represented in our multiethnic cohort of patients with SSc) is D'=0.72 (R2=0.49) and D'=0.92 (R2=0.66), respectively. We used Roadmap Epigenomics and ENCODE project data27 to search for additional evidence indicating a potential regulatory role of the GSDMA eQTL in MDMs (SNP rs3859192) and found, among other cell types, multiple overlapping regulatory marks in both primary and in CD14+ monocytes (figure 2E).
Discussion
Large-scale genetic mapping studies have yielded novel hypotheses for genes and pathways associated with SSc.3 In addition to genetic studies, assessing the specific contribution of different cell types to the pathogenesis of SSc allows to decipher the functional context where disease susceptibility genes operate and eventually prioritise specific targets for therapeutic intervention. Host genetics influence the transcriptional response in human monocytes/macrophages in a cell-specific and stimulus-specific way and is associated with disease.28 Here we identified hundreds of genes whose expression level in macrophages is associated with SSc , ,2 highlighting a disease-mediating role for this cell type2. In comparison, a similar analysis between diffuse and limited SSc yielded only seven DE genes (online supplementary figure S3), suggesting that expression changes underlying clinical SSc subtypes might be more difficult to detect in macrophages.
Our results from differential expression and eQTL analysis in SSc macrophages, when combined with genetic susceptibility (GWAS/WES), regulatory (Roadmap Epigenomics and ENCODE) and expression and eQTL data from the skin and other cell types (GTEx24), support a previously undetected role for macrophages in GSDMA overexpression in the pathogenesis of SSc. In addition, the identification of a previously unappreciated macrophage cis-eQTL in LD with the previously reported SSc risk variant in GSDMA 16 suggests that the contribution to disease of GSDMA might be exerted by macrophages. GSDMA is a member of the recently discovered gasdermin protein family. Gasdermins were previously described as regulators of cellular swelling and lysis through formation of membranous pores in conjunction with release of proinflammatory cytokines, a process also known as pyroptosis.29–31 Accordingly, Gsdma3-mutant mice with constitutive pyroptosis display severe skin inflammation.32 Thus, we speculate that overexpression of GSDMA could cause dysregulation of the pyroptosis process in SSc. Taken together our integrated expression and eQTL analyses in SSc provide a proof of concept for the functional annotation of genes that have been implicated in disease susceptibility but are poorly characterised at the cellular level, prompting detailed functional studies of immune cells in SSc.
Supplementary file 4
References
Footnotes
AM-M and MB contributed equally.
Handling editor Josef S Smolen
Contributors AM-M and MB equally contributed to this work. MB, SK and KJ-H carried out all experiments. AM-M, NH and EP designed and carried out data analyses. LG contributed to the generation of RNA-seq data. DJA, CF and CPD recruited the patients with SSc, collected samples, carried out clinical investigation and provided clinical data for the study. JM provided genetic data. AM-M, MB, JB and EP had full access to all the data in the study and take responsibility for its integrity and the data analysis. AM-M, MB, JB and EP wrote and revised the manuscript. JB and EP designed and coordinated the study.
Funding This work was supported by the Medical Research Council (MR/M004716/1 to JB and EP, and MR/N01121X/1 to JB), by the NMRC (grant CBRG15may062) and Duke-NUS Medical School (to EP), by the Arthritis Research UK (19427), Scleroderma & Raynaud’s UK and The Royal Free Charity (to DJA, VO and CPD), and by grant SAF2015 66761 P (to JM).
Competing interests None declared.
Patient consent Obtained.
Ethics approval This study was approved by the local research ethics committee (NRES Committee London Hampstead HRA reference 6398).
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The RNA-seq data generated in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) database (GEO series accession number GSE104174). Full results are provided in the online supplementary tables.