Article Text

Abstract
Objective To evaluate the similarities between LBEC0101 (etanercept biosimilar) and the etanercept reference product (ETN-RP) in terms of efficacy and safety, including immunogenicity, in patients with active rheumatoid arthritis despite methotrexate treatment.
Methods This phase III, multicentre, randomised, double-blind, parallel-group, 54-week study was conducted in Japan and Korea. The primary efficacy endpoint was the change from baseline in the disease activity score in 28 joints based on erythrocyte sedimentation rate (DAS28-ESR) at week 24. American College of Rheumatology 20% (ACR20) response rate, adverse events (AEs), pharmacokinetics and development of antidrug antibodies (ADAs) were also evaluated.
Results In total, 374 patients were randomised to LBEC0101 (n=187) or ETN-RP (n=187). The least squares mean changes from baseline in DAS28-ESR at week 24 in the per-protocol set were −3.01 (95% CI −3.198 to −2.820) in the LBEC0101 group and −2.86 (95% CI −3.051 to −2.667) in the ETN-RP group. The estimated between-group difference was −0.15 and its 95% CI was −0.377 to 0.078, which was within the prespecified equivalence margin of −0.6 to 0.6. ACR20 response rates at week 24 were similar between the groups (LBEC0101 93.3% vs ETN-RP 86.7%). The incidence of AEs up to week 54 was comparable between the groups (LBEC0101 92.0% vs ETN-RP 92.5%), although fewer patients in the LBEC0101 group (1.6%) than the ETN-RP group (9.6%) developed ADAs.
Conclusion The clinical efficacy of LBEC0101 was equivalent to that of ETN-RP. LBEC0101 was well tolerated and had a comparable safety profile to ETN-RP.
Trial registration number NCT02357069.
- Rheumatoid Arthritis
- Methotrexate
- Das28
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Etanercept (ETN) is a dimeric fusion protein produced by recombinant DNA technology in a Chinese hamster ovary mammalian expression system. It consists of disulfide bond-linked chains of a fusion protein—the soluble domain of tumour necrosis factor receptor II (TNFR II) linked to the Fc region of human immune globulin G1.1
TNF-α is considered to be a major contributor to the inflammatory response in diseases such as rheumatoid arthritis (RA) and psoriasis.2 3 The TNFR II domain of ETN binds to TNF and blocks its interaction with cell surface TNF receptors.4 Taking advantage of this activity, TNF-α inhibitors, including ETN, are used in the treatment of immune-related inflammatory diseases.5 6 In Japan, etanercept reference product (ETN-RP) was approved for the treatment of RA (including the prevention of structural damage of joints) and polyarticular-course juvenile idiopathic arthritis, in patients with inadequate response to conventional therapy. In Korea, ETN-RP was approved for the treatment of RA, psoriatic arthritis, axial spondyloarthritis (ankylosing spondylitis) and psoriasis in adults, and juvenile idiopathic arthritis in paediatric patients.
A biosimilar is a biological medicinal product that contains a version of the active substance of an already authorised original biological medicinal product (reference medicinal product).7 LBEC0101 has been developed as a biosimilar product to ETN-RP. The high similarity in the structural and functional properties and biological activities between LBEC0101 and ETN-RP has been demonstrated by in vitro and in vivo studies such as a TNF-α binding affinity study. In addition, for the clinical development of LBEC0101, a phase I pharmacokinetic (PK) study in healthy male volunteers was conducted and showed equivalent PKs between single-dose LBEC0101 and ETN-RP.8
The objective of the present study was to compare the efficacy and safety of LBEC0101 and ETN-RP as adjunctive therapy to methotrexate (MTX) in patients with active RA and poor responses to previous MTX treatment. As part of the safety evaluation, immunogenicity was also evaluated.
Methods
Participants
Patients aged 20–75 years who had been diagnosed with RA for ≥6 months were recruited. At screening, patients were required to meet the 1987 revised American College of Rheumatology (ACR) classification criteria,9 to be classified as functional class I, II or III according to the 1991 revised ACR criteria10 and to have active RA (≥6 swollen joints, ≥6 tender joints, erythrocyte sedimentation rate ≥28 mm/hour or serum C reactive protein ≥1.0 mg/dL and disease activity score in 28 joints based on erythrocyte sedimentation rate (DAS28-ESR) ≥3.2) despite MTX treatment for ≥12 weeks, including 4 weeks of receiving a stable MTX dose before randomisation. Patients were excluded if they had active tuberculosis at screening, although patients with latent tuberculosis could participate under the condition of isoniazid treatment for ≥3 weeks prior to the first administration of the study drugs. Patients were also excluded if they had ever received ≥2 biological therapies for RA or previously received ETN treatment.
Study design and treatment
This phase III, multicentre, randomised, double-blind, parallel-group, 54-week study was conducted at 30 centres in Korea and 48 centres in Japan. After the screening period, patients were randomly assigned to receive 50 mg of either LBEC0101 or ETN-RP subcutaneously once a week for 52 weeks, followed by a post-treatment follow-up period of 2 weeks. The study design is shown in online supplementary figure S1. Throughout the entire study period, MTX was coadministered to all patients on a stable dose (7.5–15 mg/week in Korea and 6–16 mg/week in Japan, based on the approved dose in each country). Allowed concomitant medications were non-steroidal anti-inflammatory drugs, analgesic drugs and oral/suppository/topical/bronchial/nasal corticosteroids (≤10 mg/day prednisone equivalent dose). The doses of these medications were not allowed to be changed from 4 weeks before the first study drug administration. Prohibited concomitant medications included disease-modifying antirheumatic drugs (DMARDs) other than MTX and intravenous/intramuscular/intra-articular/epidural corticosteroids.
Supplementary file 1
Randomisation and blinding
Randomisation was performed centrally using an interactive web/voice response system. After screening, patients who met all eligibility criteria were randomly assigned to one of the two treatment groups in a 1:1 ratio. The randomisation scheme ensured balance between the groups, using the status of previous use of a biological DMARD for RA and country as stratification factors. Study drug packaging was double-blinded throughout the study.
When all enrolled patients completed week 24 assessments or discontinued prior to week 24, the database was locked and unblinded data were disclosed to personnel involved in the statistical analysis and medical writing only for marketing authorisation applications. The blind-to-treatment allocation was strictly maintained for the remainder of study period among all patients, investigators and applicable study staff to prevent evaluation bias. The procedures for unblinding and maintenance of blinding were predesignated.
Efficacy
The primary endpoint was the change from baseline in DAS28-ESR at week 24. The secondary efficacy endpoints were the changes from baseline in DAS28-ESR at weeks 12 and 52 and ACR20 response rates at weeks 12, 24 and 52. Other efficacy endpoints included ACR50, ACR70 and European League Against Rheumatism (EULAR) response rates based on the DAS28-ESR at weeks 12, 24 and 52. The EULAR response criteria were defined as no response, moderate response or good response based on the present DAS28-ESR and improvement in the DAS28-ESR from baseline.
Safety
Safety endpoints included incidence of adverse events (AEs) and serious adverse events (SAEs) up to week 54. AEs of special interest that were known as key safety issues for ETN-RP, such as infections, sepsis, injection site reactions, malignancies, heart failure, neurological events, tuberculosis, hepatitis B (HBV) reactivation and interstitial lung disease, were also investigated. AEs were coded using MedDRA V.19.0.
Pharmacokinetics
Serum trough concentrations (Ctrough) of ETN at weeks 12, 24 and 52 were analysed by a validated immunofluorescence assay using Gyrolab platform (Gyros AB, Uppsala, Sweden).
Immunogenicity
Antidrug antibodies (ADAs) and neutralising antibodies at weeks 0, 12, 24 and 52 were analysed by validated electrochemiluminescent immunoassay using Meso Scale Discovery platform (Meso Scale Diagnostics, Rockville, Maryland, USA). Biotinylated ETN-RP and SULFO-TAG labelled ETN-RP were used to detect ADAs, and biotinylated ETN-RP and SULFO-TAG labelled TNF-α were used to detect neutralising antibodies. Neutralising antibody tests were performed only when the results were positive for ADAs.
Statistical analysis
Based on previous studies comparing DAS28-ESR between an ETN-RP and MTX combination treatment group and an MTX monotherapy group, it was expected that the effect size would be 1.4.11 12 A change in DAS28-ESR of more than 0.6 from baseline was considered clinically significant according to the EULAR response criteria, which define a DAS28 improvement of 0.6 or less as no response to treatment. Therefore, the equivalence margin for the similarity in change from baseline in DAS28-ESR at week 24 between the groups was set as −0.6 to 0.6 in this study. A sample size of 372 patients (186 per treatment group, 1:1 ratio) for randomisation was estimated to demonstrate equivalence between the groups for the primary efficacy endpoint, change from baseline in DAS28-ESR at week 24, with two one-sided tests at an alpha level of 2.5% and 90% statistical power. It was assumed that the between-group difference was 0.0, the SD in each treatment group was 1.4 and the per-protocol set (PPS) dropout rate was 20%.
Efficacy at weeks 12 and 24 was analysed using the PPS for data up to week 24 (PPS-24w), and efficacy at week 52 was analysed using the PPS for data up to week 52 (PPS-52w). The full analysis set (FAS) was used for checking the sensitivity of the PPS results. Detailed FAS and PPS criteria are shown in the online supplementary text.
The point estimate of the treatment difference and CI of the DAS28-ESR change were derived using analysis of covariance (ANCOVA). The ANCOVA model included independent variables of the fixed categorical effects of treatment, country of the study centre, status of previous use of a biological DMARD and the baseline DAS28-ESR as a covariate. For the PPS analysis, missing data were not replaced. The FAS analysis was performed using imputation of missing data with the post-baseline last observation carried forward method. All safety data, including immunogenicity data, were analysed using the safety analysis set (SS), which included all patients who received at least one dose of the study drug and summarised by treatment group using descriptive statistics. The Ctrough was analysed using the PK analysis set (PKS), which included all patients who had at least one serum Ctrough measurement after study drug administration, and summarised using descriptive statistics such as the geometric mean of each treatment. The analysis was performed using SAS V.9.3 (SAS Institute Inc., Cary, North Carolina, USA).
Results
Patient disposition and baseline characteristics
This study was conducted between February 2015 and December 2016. The disposition of patients is shown in figure 1. Of the 517 patients screened, 187 subjects were randomised in each treatment group. Twenty-eight patients in the LBEC0101 group and 24 patients in the ETN-RP group discontinued the study—most commonly because of AEs (LBEC0101 n=11, ETN-RP n=13) and withdrawal of consent (LBEC0101 n=11, ETN-RP n=7). In total, 159 and 163 patients in the LBEC0101 and ETN-RP groups, respectively, completed the entire treatment period, up to week 52.

Patient disposition. A total of 517 patients were screened and 374 patients were randomised. Almost equal numbers of patients in Japan and Korea were allocated to each treatment group. Period I and period II mean weeks 0–24 and weeks 24–52, respectively. AEs, adverse events; ETN-RP, etanercept reference product.
Data were obtained for 374 randomised patients. Of these, data for two patients were excluded from the FAS (n=372) because they had no DAS28-ESR assessment after baseline. Data for 45 and 78 patients were excluded from the PPS-24w (n=329) and the PPS-52w (n=296), respectively, because they had no DAS28-ESR assessments at week 24 and/or week 52 or they did not meet other criteria of the PPS-24w or PPS-52w. No patients were excluded from the SS (n=374), and two patients were excluded from the PKS (n=372) because of missing serum Ctrough measurements after study drug administration.
The demographic characteristics of the patients and the patients’ use of concomitant medications in the FAS are summarised in table 1. There were no major differences in demographic characteristics between the groups. The proportions of patients who used one biological DMARD prior to study participation and who used corticosteroids at baseline were also similar between the groups.
Baseline patient demographic and disease characteristics (full analysis set)
Efficacy
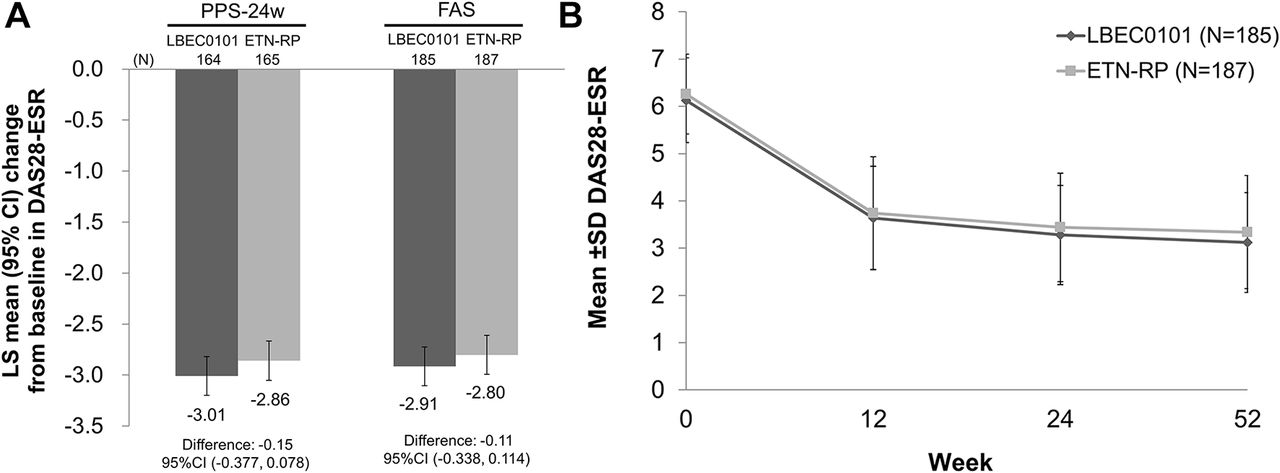
The least squares mean changes from baseline in DAS28-ESR at week 24, the primary efficacy endpoint, were −3.01 (95% CI −3.198 to −2.820) in the LBEC0101 group and −2.86 (95% CI −3.051 to −2.667) in the ETN-RP group (figure 2A). The estimated between-group difference was −0.15 and its 95% CI was −0.377 to 0.078, which was within the prespecified equivalence margin (−0.6 to 0.6), indicating equivalence in efficacy between LBEC0101 and ETN-RP. A similar result was found in the FAS (−0.11 (95% CI −0.338 to 0.114)).

DAS28-ESR change from baseline. (A) LS mean change from baseline in DAS28-ESR at week 24 and its 95% CI adjusted for the country of the study centre, status of previous use of a biological disease-modifying antirheumatic drugs and DAS28-ESR at baseline (PPS-24w and FAS). (B) Mean±SD of DAS28-ESR at baseline, week 12, week 24 and week 52 (FAS). DAS28-ESR, disease activity score in 28 joints based on erythrocyte sedimentation rate; ETN-RP, etanercept reference product; FAS, full analysis set; LS, least squares; PPS-24w, per-protocol set for data up to week 24.
The mean DAS28-ESR rapidly declined up to week 12 and almost plateaued after week 24 in both groups (figure 2B). A subgroup analysis demonstrated that both study drugs were effective, regardless of country, previous use of biological DMARD, baseline DAS28-ESR and baseline MTX dose (online supplementary table S1). The percentages of patients achieving remission (DAS28-ESR <2.6) and low disease activity (DAS28-ESR ≤3.2) are shown in online supplementary figure S2.
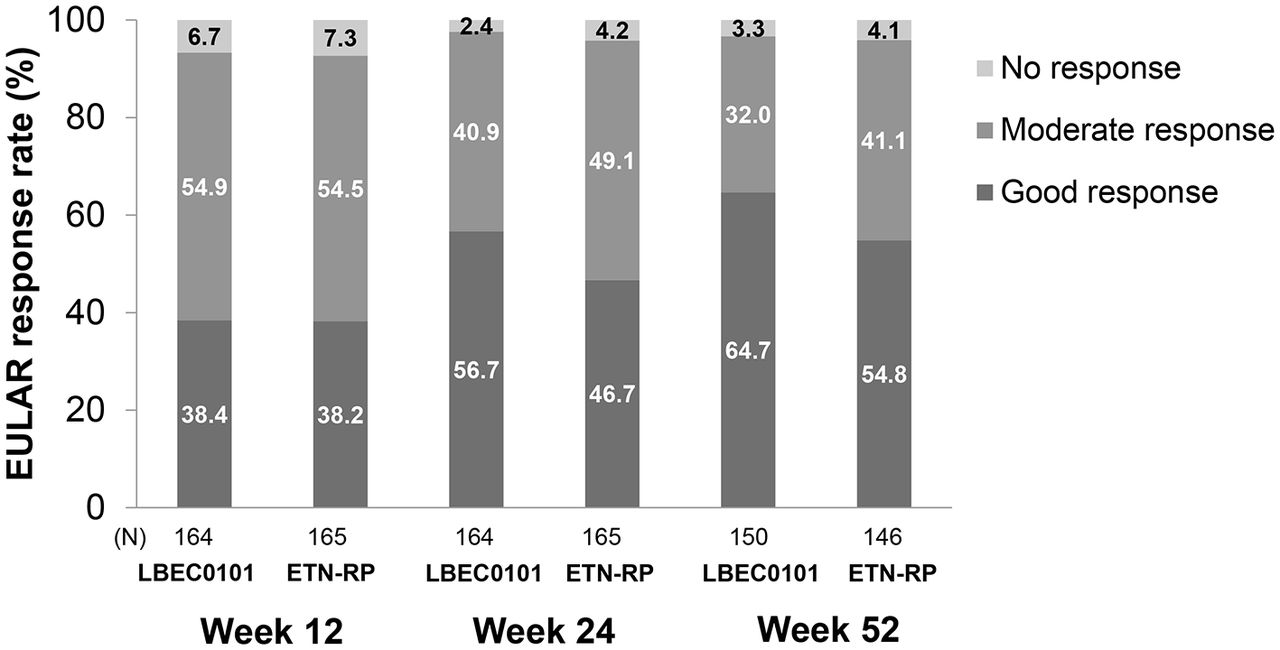
ACR20 response rates, the secondary efficacy endpoint, were 93.3% in the LBEC0101 group and 86.7% in the ETN-RP group at week 24. The between-group difference in ACR20 (95% CI) was 6.6% (0.2 to 13.1) in the PPS-24w (figure 3A). The ACR50 and ACR70 response rates also showed similar trends to the ACR20 response rate (figure 3B,C). Figure 4 shows more than 90% of patients achieved a moderate to good EULAR response at weeks 12, 24 and 52, and after week 24, less than 5% of patients remained as non-responders in both groups.

ACR20 (A), ACR50 (B), and ACR70 (C) response rates at weeks 12 and 24 (PPS-24w) and at week 52 (PPS-52w). ACR, American College of Rheumatology; ETN-RP, etanercept reference product; PPS-24w, per-protocol set at 24 weeks; PPS-52w, per-protocol set at 52 weeks.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
EULAR response at weeks 12 and 24 (PPS-24w) and at week 52 (PPS-52w) ETN-RP, etanercept reference product; EULAR, European League Against Rheumatism; PPS-24w, per-protocol set at 24 weeks; PPS-52w, per-protocol set at 52 weeks.
Safety
In total, 172 (92.0%) and 173 (92.5%) patients developed AEs in the LBEC0101 and ETN-RP groups, respectively. In the LBEC0101 group, 44 SAEs, including three deaths, were reported in 31 (16.6%) patients. In the ETN-RP group, 28 SAEs, including one death, were reported in 20 (10.7%) patients. The causes of death in the LBEC0101 group were circulatory failure, acute heart failure and acute respiratory distress syndrome; the causality relationship between these events and the study drug could not be ruled out. The one case of death in the ETN-RP group was suicide, which was not considered related to the study drug. The incidence of SAEs in the LBEC0101 group (16.6%) was higher than that in the ETN-RP group (10.7%) because there were more events unrelated to the study drugs, such as traffic accident injury, accidental falls and events caused by concomitant diseases, in the LBEC0101 group. The incidence of serious related AEs was the same in both groups (7.0%).
The most frequently reported AEs are shown in table 2. Most of the AEs were mild in severity.
AEs during the entire study period and AEs experienced by >5% of patients in either group during the entire study period
Regarding AEs of special interest, no difference in the incidence of infection between the groups was found (LBEC0101 54.5% vs ETN-RP 54.5%), although three patients in each group experienced interstitial lung disease and only one patient in the LBEC0101 group experienced sepsis. Injection site reactions occurred in fewer patients and less frequently in the LBEC0101 group than in the ETN-RP group (LBEC0101 10.2% (19 patients, 77 events) vs ETN-RP 34.2% (64 patients, 438 events)). Most of these events occurred early, before week 12, and were mild in severity in both treatment groups. Malignancies occurred in four patients in the ETN-RP group only and heart failure occurred in one patient (0.5%) in the LBEC0101 group only. No neurological events, active tuberculosis or occurrences of HBV reactivation were reported in either treatment group.
Pharmacokinetics
The mean Ctrough values between week 12 and week 52 ranged from 2579.1 ng/mL to 4143.4 ng/mL in the LBEC0101 group, and from 2166.8 ng/mL to 3586.5 ng/mL in the ETN-RP group (online supplementary figure S3).
Immunogenicity
Two (1.1%) patients in the ETN-RP group were ADA-positive at baseline. After treatment, three (1.6%) patients in the LBEC0101 group and 18 (9.6%) patients in the ETN-RP group developed ADAs (online supplementary table S2). None of them was positive for neutralising antibody. The effect of ADAs on PK, efficacy and safety was difficult to determine due to the small number of ADA-positive patients in the LBEC0101 group. No ADA-positive patients had an injection site reaction in the LBEC0101 group, although the incidence of injection site reaction was 10.2% in the overall LBEC0101 group.
Discussion
This phase III study was designed to evaluate the similarities between LBEC0101 and ETN-RP, in terms of efficacy, safety and immunogenicity, in patients with active RA who had an inadequate response to MTX. Based on the prespecified equivalence margin of −0.6 to 0.6, the equivalence of LBEC0101 to ETN-RP was confirmed in terms of the primary efficacy endpoint, the change in DAS28-ESR at week 24. This study is considered to have had an appropriate assay sensitivity because it had a similar study design and results to previous studies that demonstrated superior efficacy of MTX and ETN-RP combination therapy over MTX monotherapy.
Although many previous ETN-RP studies and other ETN biosimilar studies used ACR20 as the primary endpoint, this study adopted DAS28-ESR as the primary endpoint. Given that the regulatory guidance stating that continuous variables such as DAS28 may be more sensitive to change13 and that the most sensitive clinical endpoint is preferred for detecting product-related differences for biosimilars,14 DAS28-ESR was considered an appropriate primary endpoint for a biosimilar study.
The results of ACR20 response rate at weeks 12, 24 and 52 also supported the similarity between LBEC0101 and ETN-RP. In this study, the ACR20 response rates in the LBEC0101 and ETN-RP groups at week 24 (93.3% and 86.7%, respectively) were slightly higher than what was reported in previous clinical studies for ETN-RP.15–17 We suppose that higher ACR20 response rates can be observed in active-comparator studies because participants and investigators would have known that the active drug was used in both groups, and therefore expected the study drugs to be effective. Previous biosimilar studies showed the same trends18–20 in comparison with placebo-controlled studies for innovator drugs.
The safety profile of LBEC0101 was almost comparable with that of ETN-RP, although fewer injection site reactions and fewer ADAs were observed in the LBEC0101 group. The reason for the differences in incidence of injection site reactions and ADAs between the groups has not been identified yet. Given that the ADA assay system adopted in this study used labelled ETN-RP, another assay system utilising labelled LBEC0101 was also developed and all the samples were measured. However, the assay system utilising labelled LBEC0101 demonstrated a similar result (LBEC0101: 3.7% vs ETN-RP: 15.0%), suggesting that the difference in ADA incidence is not ascribed to the assay system. Previous studies comparing other ETN biosimilars, SB4 and GP2015, with ETN-RP reported similarly lower incidences of injection site reactions and ADAs in biosimilar groups.19 21 The study report suggested that differences in the formulation and container closure system might have contributed to the lower incidence of injection site reactions, and that product aggregates, impurities and container closure system might have contributed to the lower incidence of ADAs in the biosimilar treatment group, although the true causes were uncertain.19 Similar reasons may be considered in the present study.
The mean Ctrough value of LBEC0101 was relatively higher than that of ETN-RP; however, the high interpatient variability may have contributed to the difference.
Because the present study was the first clinical trial of LBEC0101 for RA conducted in Asia (Japan and Korea) only, the generalisability of the findings may be limited to the Asian population and a longer term study is warranted. However, based on the known ETN-RP properties and the high similarity between LBEC0101 and ETN-RP, LBEC0101 is very likely to be insensitive to both intrinsic and extrinsic ethnic factors. In addition, long-term evaluations over 52 weeks and switch to LBEC0101 from ETN-RP are currently being explored in an extension study (an additional 48 weeks after the 52-week treatment period in the present study) to evaluate the long-term safety, efficacy and immunogenicity o LBEC0101.
In conclusion, LBEC0101 was shown to be equivalent to ETN-RP in terms of clinical efficacy. LBEC0101 was well tolerated with a comparable safety profile to ETN-RP.
Acknowledgments
The authors would like to thank all the patients who participated in this study and all the principal investigators: Kanzo Amano, Masato Araki, Kohei Azuma, Nobutaka Eiraku, Hideo Hashimoto, Masatoshi Hayashi, Wataru Hirose, Tomomaro Izumihara, Satoshi Jodo, Motohide Kaneko, Kou Katayama, Atsushi Kawakami, Mitsuyo Kinjo, Shigehito Kiyokawa, Yoshinobu Koyama, Tsukasa Matsubara, Hajime Miyasato, Atsuo Nakajima, Hiroshi Nakamura, Tadashi Nakamura, Kusuki Nishioka, Susumu Nishiyama, Junichi Obata, Shuji Ohno, Hideo Ohtsubo, Yutaka Okano, Ichiro Oki, Motohiro Oribe, Kei Osano, Takaaki Sagara, Akira Sagawa, Kenmei Sakata, Ichiro Seike, Koichi Shigenobu, Eisuke Shono, Yuichi Takahashi, Koichiro Tanaka, Masahiro Tanaka, Kazuhide Tanimura, Kiyoyuki Tokuyama, Seiji Tsuboi, Shoji Uchida, Eishi Uechi, Akihiro Yamaguchi, Yuji Yamanishi, Akira Yokota, Masaaki Yoshida, Tomohiko Yoshida, Tamami Yoshitama, Seiji Yoshizawa, Han Joo Baek, Hoon-Suk Cha, Jung-Yoon Choe, Chan-Bum Choi, In Ah Choi, Seung-Jae Hong, Jin-Wuk Hur, Geun-Tae Kim, Hyun Ah Kim, Jinseok Kim, Jisoo Lee, Sang-Heon Lee, Sang-Il Lee, Seung-Geun Lee, Shin-Seok Lee, Yun Jong Lee, Jun-Ki Min, Kyung-Su Park, Min-Chan Park, Sung-Hwan Park, Won Park, Yong-Beom Park, Dong Hyuk Sheen, Seung Cheol Shim, Kichul Shin, Chang-Hee Suh, Hyung-In Yang, Bin Yoo and Wan-Hee Yoo.
References
Footnotes
Handling editor Tore K Kvien
Contributors All authors (1) were involved in the conception and design of the study, or the acquisition, analysis and interpretation of data for the study, (2) were involved in drafting the manuscript and revising it critically for important intellectual content, (3) gave final approval of the version to be published, and (4) agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This study was funded by LG Chem (formerly LG Life Sciences), Mochida Pharmaceutical and Korea Health Industry Development Institute. Medical writing and editorial assistance, funded by Mochida Pharmaceutical, was provided by Michelle Belanger, MD, of Edanz Medical Writing.
Competing interests HM has received consulting fees for this study from Mochida Pharmaceutical, consulting fees unrelated to this study from AYUMI Pharmaceutical Corporation, Nichi-Iko Pharmaceutical and Meiji Seika Pharma, and lecture fees from Daiichi Sankyo, UCB Japan, Janssen Pharmaceutical KK, Chugai Pharmaceutical and Ono Pharmaceutical. MT and AH are employees of Mochida Pharmaceutical. SS and JL are employees of LG Chem. YWS received a grant for this study from LG Chem.
Patient consent Detail has been removed from this case description/these case descriptions to ensure anonymity. The editors and reviewers have seen the detailed information available and are satisfied that the information backs up the case the authors are making.
Ethics approval This study was conducted according to the Declaration of Helsinki, International Committee on Harmonization Good Clinical Practice guideline, and applicable local laws and regulations. The protocol was approved by the regulatory authorities in each country and by the ethics committees of each study site.
Provenance and peer review Not commissioned; externally peer reviewed.
Presented at Part of this manuscript was presented at the 2017 ACR/ARHP Annual Meeting.