Article Text

Abstract
Objectives Chondroitin sulfate 800 mg/day (CS) pharmaceutical-grade in the management of symptomatic knee osteoarthritis consistent with the European Medicines Agency guideline.
Methods A prospective, randomised, 6-month, 3-arm, double-blind, double-dummy, placebo and celecoxib (200 mg/day)-controlled trial assessing changes in pain on a Visual Analogue Scale (VAS) and in the Lequesne Index (LI) as coprimary endpoints. Minimal-Clinically Important Improvement (MCII), Patient-Acceptable Symptoms State (PASS) were used as secondary endpoints.
Results 604 patients (knee osteoarthritis) diagnosed according to American College of Rheumalogy (ACR) criteria, recruited in five European countries and followed for 182 days. CS and celecoxib showed a greater significant reduction in pain and LI than placebo. In the intention-to-treat (ITT) population, pain reduction in VAS at day 182 in the CS group (−42.6 mm) and in celecoxib group (−39.5 mm) was significantly greater than the placebo group (−33.3 mm) (p=0.001 for CS and p=0.009 for celecoxib), while no difference observed between CS and celecoxib. Similar trend for the LI, as reduction in this metric in the CS group (−4.7) and celecoxib group (−4.6) was significantly greater than the placebo group (−3.7) (p=0.023 for CS and p=0.015 for celecoxib), no difference was observed between CS and celecoxib. Both secondary endpoints (MCII and PASS) at day 182 improved significantly in the CS and celecoxib groups. All treatments demonstrated excellent safety profiles.
Conclusion A 800 mg/day pharmaceutical-grade CS is superior to placebo and similar to celecoxib in reducing pain and improving function over 6 months in symptomatic knee osteoarthritis (OA) patients. This formulation of CS should be considered a first-line treatment in the medical management of knee OA.
- knee osteoarthritis
- treatment
- chondroitin sulfate
- pain
- function
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA) is the most prevalent musculoskeletal disease affecting humans, an important cause of pain, loss of function, disability and a major public health problem1 2 that is associated with a substantial and ever increasing burden on society.3 4 OA of the knee and hip tends to generate the greatest impact on the population, as pain and stiffness in these large weight-bearing joints often lead to the need for medical intervention.2 Medical management of knee OA includes both pharmacological and non-pharmacological modalities and numerous scientific societies have produced recommendations for the non-surgical management of knee OA.5–9 Although several differences are observed between these evidence-based guidelines, mostly reflecting heterogeneity of the expert panels involved, geographical differences in the availability of chemical entities 10 11 there was, until recently, a general consensus that analgesics, including paracetamol and non-steroidal anti-inflammatory drugs (NSAIDs) have demonstrated a positive benefit-risk profile when used to treat symptoms of knee OA.5–11 However, recent publications have aggressively challenged the use of paracetamol for the treatment of symptomatic OA because of a lack of efficacy and a considerable degree of toxicity, especially at the upper end of the standard analgesic dose.12–14 Similarly, safety profiles of oral NSAIDs remain a concern and caution is recommended before selecting the preparation and dose for a patient.14 Therefore, recent guidelines recommend maintenance therapy to be conducted with symptomatic slow-acting drugs for OA (SYSADOAs), a class of drugs that is recognised to offer a high degree of safety and tolerability.5 Although discrepancies can be found in the literature regarding recommendations on SYSADOAs in the management of knee OA,10 11 higher quality evidence seems to be provided for patented, prescription formulations of chondroitin sulfate (CS) and crystalline glucosamine sulfate (GS).9
Chondroitin sulfate (CS) is a sulfated glycosaminoglycan composed of chains of alternating D-glucuronic acid and N-acetyl-D-galactosamine.15 CS is available as pharmaceutical-grade and nutraceutical-grade products, the latter exhibiting striking variations in preparation, composition, purity as well as clinical effects. These differences may explain why, whereas pharmaceutical-grade CS (ie, the 4&6isomer of sodium CS) was shown to improve pain and function and/or delay structural progression of knee OA in several well-conducted studies,16–18 these results were not confirmed when lower grade formulations were used.19 20 Indeed, a recent systematic review conducted by the Cochrane Collaborative Group concludes that CS, alone or in combination with GS, is better than placebo in improving pain in participants with OA in short-term studies, with CS having a lower risk of serious adverse events compared with controls.21
Another potential source of inconsistency in the results from previous studies of SYSADOAs in knee OA has been idiosyncratic trial design. For this reason, the European Medicines Agency (EMA) produced a Guideline on Clinical Investigation of Medicinal Products Used in the Treatment of Osteoarthritis (CPMP/EWP/784/97 Rev. 1), guideline which has been recently supported by a European experts consensus.22 It recommends that efficacy of chemical entities used in the treatment of symptomatic OA be tested according to a standard study design with the following basic parameters: a minimum 6-month study duration; a three-arm study design including a placebo and an active comparator (ie, oral NSAID); and two co-primary endpoints evaluating pain and function, respectively.
Herein, we report results from a study of pharmaceutical-grade CS in patients with symptomatic knee OA, which, to our knowledge, is the first ever to have been conducted in full accordance with the aforementioned EMA guideline.
Material and methods
Study design and selection of patients
The study comprised patients from Belgium, Czech Republic, Italy, Poland and Switzerland, who were enrolled between June 2014 and October 2015. The main inclusion criteria were outpatients status, age above 50 years and primary knee OA of the medial or lateral femorotibial compartment diagnosed according to the clinical and radiographic criteria of the American College of Rheumatology (ACR).23 The more symptomatic knee (with a pain score of at least 50 mm on a 0–100 mm Visual Analogue Scale (VAS) for at least 3 months before enrolment) was defined as the target knee. The main exclusion criteria were those listed in the last version of the Guideline on Clinical Investigation of Medicinal Products used in the Treatment of Osteoarthritis released by the EMA in 2010 (CPMP/EWP/784/97 Rev. 1) and grade 4 radiographic OA according to the Kellgren-Lawrence (K-L) grading system.24 Use of any intra-articular injection in the target knee in the last 6 months, SYSADOAs in the last 3 months, NSAIDs in the last 5 days and paracetamol in the 10 hours preceding enrollment was also specifically forbidden by the study protocol. There were two co-primary endpoints, predefined as stipulated by the EMA guidelines: pain and Lequesne Index (LI) assessment. Ethics Committee approval from all participating centres was obtained and all patients gave their written informed consent to participate.
This study has been designed to assess the symptomatic effect of CS. Bone and cartilage markers were not the target in this short study.
Treatment assignment
Patients were randomly assigned to one of the following three groups: (1) Group CS: one tablet of CS 800 mg and one capsule of placebo celecoxib; (2) Group celecoxib: one tablet of placebo CS and one capsule of celecoxib 200 mg (Celebrex Pfizer); (3) Group placebo: one tablet of placebo CS and one capsule of placebo celecoxib. The tablets of Celebrex available on the market were encapsulated to allow for a double-blind, double-dummy design. CS tablets contained highly purified chondroitin 4 & 6 sulfate in a concentration not less than 95% (European patents E 1582214 and EP 1705192) (Condrosulf (other brand name: Chondrosulf, Condral) 800; IBSA Institut Biochimique SA; Pambio-Noranco, Switzerland). All treatments were taken once daily, every evening with a glass of water, for 6 months. For rescue analgesia, patients were allowed to take paracetamol 500 mg tablets (maximum dosage 3 g/day), and they recorded use thereof in a diary. An appropriate washout period of 10 hours was required before symptom assessment at in-clinic visits. No other pharmacological or non-pharmacological interventions for OA were allowed. Compliance with the study treatments was established by asking the patients about missing doses and by counting unused study drugs.
Outcome measures
There were two co-primary endpoints, as stipulated by the EMA guideline, and both were assessed as the change from baseline, that is, the difference between enrollment and study conclusion. One endpoint was the patient’s estimate of pain on a 100 mm Visual Analogue Scale (VAS). The other endpoint was the Lequesne Index (LI), which integrates pain and function and results in a score from 0 to 24.25 Secondary endpoints included the proportion of patients reaching the Minimal-Clinically Important Improvement (MCII), defined as the smallest change in measurement that signifies an important improvement in a patient’s symptom,26 and the Patient Acceptable Symptom State (PASS), defined as the value of symptoms beyond which patients consider themselves well.27 Patient and investigator global assessment were scored on a 5-point Likert ordinal scale (excellent, good, fair, poor, none). All adverse events and abnormal laboratory test results were recorded.
Statistical analysis
All statistical analyses were performed using SAS Software (V. 9.4 and V. 8.2) on a Windows 7 operating system.
We calculated a sample size of 600 patients based on an estimated difference of 9 mm between CS and placebo after 6 months of treatment, with a standard deviation (SD) of 25 mm, a power of 90%, an alpha risk of 5% and a drop-out rate of 15%. The intention-to-treat (ITT) population was defined as all randomised patients who received one dose of the study medication. Safety analyses were conducted on all randomised patients.
VAS (pain in mm) and LI score from D1 to D182 were compared between the three treatment groups by means of a linear mixed model carried out by using the SAS MIXED procedure, with patient as random effect, centre, treatment group, time point, interaction between treatment group and time point as categorical covariates (interaction between treatment group and centre excluded from the final models because not statistically significant. p=0.101 for VAS mixed model, p=0.998 for LI mixed model). No missing values replacement (LOCF, last observation carried forward or BOCF, basal observation carried forward) was performed for this analysis. The proportion of patients reaching the MCII, the PASS and the Outcome Measures in Rheumatology (OMERACT-OARSI) criteria were compared using a Chi-square (χ2)) test. Patient’s and investigator’s global assessments were analysed by means of Mantel-Haenszel χ2 test. Differences between groups in rates of patients with treatment-emergent adverse events (TEAEs), serious adverse events (SAEs), adverse drug reactions (ADRs) and study withdrawals due to AEs were assessed using the χ2 test.
Results
Of 656 patients screened, 604 were randomised and 603 considered eligible for ITT analysis (all patients who received the study medication). Of these patients, 199 received CS, 199 received celecoxib and 205 received placebo. The cumulative time distribution of withdrawals was similar in the three groups without significant differences in reasons for withdrawals (figure 1). Patients in the three groups had similar demographic and baseline characteristics (table 1). Grades 1 to 3 of K-L were equally distributed between the three groups, with roughly 50% of the patients presenting a grade 2 OA and 25% corresponding to either a grade 1 or a grade 3 overall.
Demographic and baseline characteristics of patients
VAS and LI

Disposition of patients. AE, adverse events.
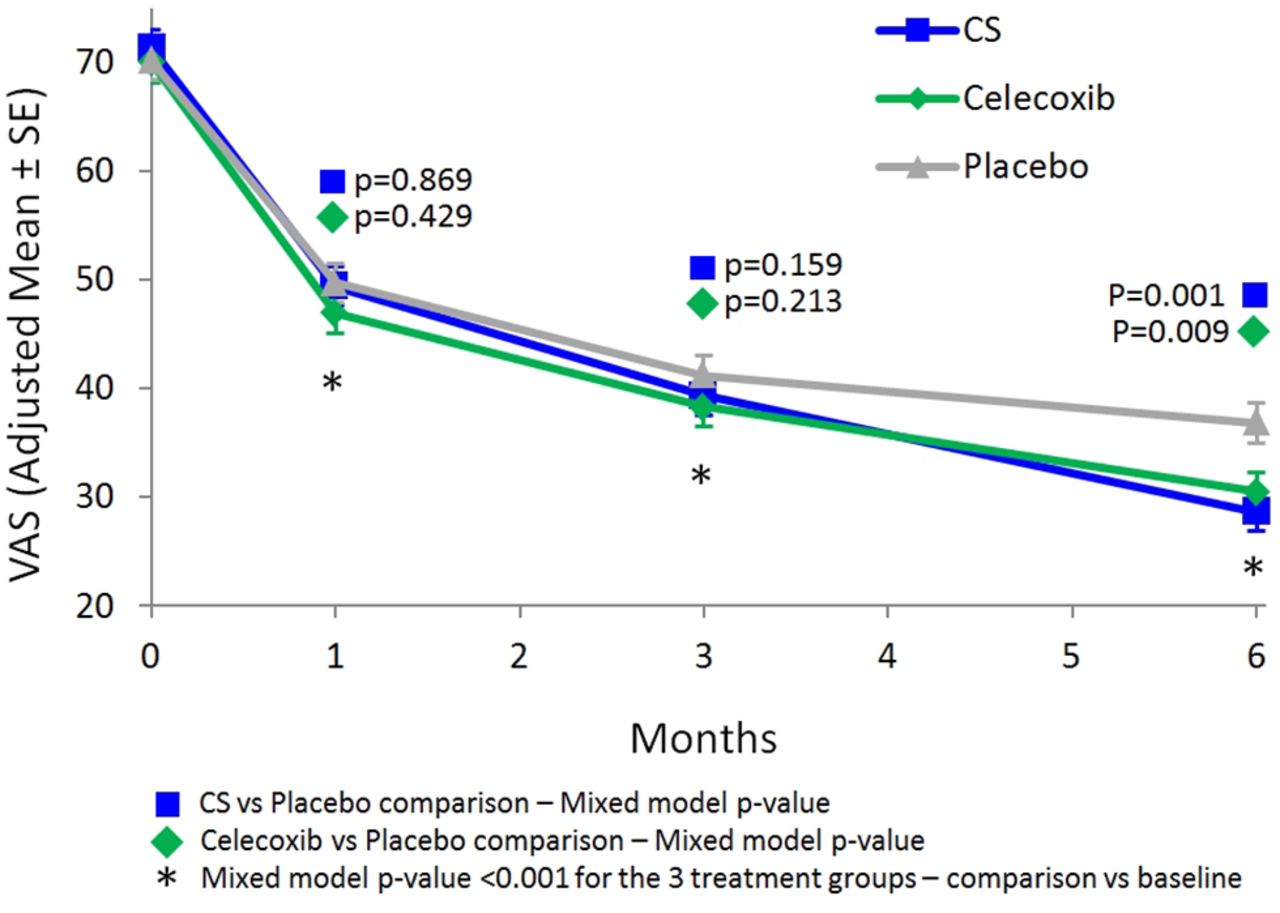
Analysis of pain scores in ITT revealed a significant improvement in all three groups compared with baseline at day 30, 91 and 182 (all p<0.001) (figure 2). Both the CS and the celecoxib group showed a statistically greater reduction in pain compared with the placebo group (p=0.001 for CS and p=0.009 for celecoxib, table 2) after 6 months without any significant difference between the two active groups (p=0.446).

Visual Analogue Scale (VAS).
Analysis of LI scores in ITT revealed a significant amelioration in all three groups compared with baseline at day 30, 91 and 182 (all p<0.001) (figure 3). At day 91 and 182, both CS and celecoxib induced a significantly greater reduction in LI than placebo (p=0.050 for CS and p=0.027 for celecoxib at day 91, p=0.023 for CS and p=0.015 for celecoxib at day 182) while no difference was observed between CS and celecoxib (p=0.799 at day 91 and p=0.890 at day 182, table 2). The decrease in LI observed in the celecoxib group attained statistical significance in comparison to the placebo group at day 30 (p=0.045), while it took the CS group until day 91 (p=0.050) (figure 3).

{kind=link}
{kind=link}
{kind=link}
Lequesne Index (LI).
After 6 months, a greater proportion of patients reached the MCII (20 mm of VAS reduction) in the CS (68%) and celecoxib (69%) groups than in the placebo group (61%). This difference was not significant for the CS–placebo comparison (p=0.122), for the celecoxib–placebo comparison (p=0.098) and not significant for CS–celecoxib comparison (p=0.914). Similar results were obtained for the proportion of patients reaching the PASS in the CS (57%), celecoxib (59%) and placebo (49%) groups. The PASS data were significant for the celecoxib–placebo comparison (p=0.047), not significant for the CS–placebo comparison (p=0.130) and not significant for the CS–celecoxib comparison (p=0.611).
Significant results were observed when defining responders patients with at least 40% or 50% of improvement in pain or LI scores, and when patients were classified according to OMERACT-OARSI (scenario F). CS and celecoxib provided significantly higher number of responders than placebo and no difference was observed between CS and celecoxib (table 3).
OMERACT-OARSI and MCII
At study conclusion (day 182), significantly more patients and more investigators scored the global assessment as excellent or good in the CS and celecoxib groups compared with the placebo (p=0.027 for CS, p=0.013 for celecoxib), while there was no difference between the two active groups (p=0.774). Study medication usage was >95% in all groups, demonstrating excellent compliance and the absence of intergroup differences.
Finally, there was no significant difference between CS, celecoxib or placebo usage in the rate of TEAEs, SAEs, ADRs and withdrawal related to TEAEs. Abdominal pain/discomfort was the most frequently reported ADR (2.5% in the CS group, 4.5% in the celecoxib group and 2.9% in the placebo group). Routine laboratory testing identified one case of leukopenia and one case of thrombocytopenia in the placebo group, but no significant abnormalities in the CS or celecoxib groups.
Discussion
In this report, we provide data from the CONCEPT trial, which, to our knowledge, is the first-ever evidence supporting a durable therapeutic benefit of SYSADOAs in a knee OA clinical trial that is fully aligned with the current EMA guideline. We demonstrated that CS is superior to placebo and similar to celecoxib across multiple outcome measures, including reduction in pain and LI (co-primary endpoints), as well as in the proportion of patients experiencing MCII (secondary endpoint) and patient/investigator global assessments.
Prior to CONCEPT, the only study that assessed the impact of a SYSADOA on knee OA in a three-arm design was the Glucosamine Unum-in-Die Efficacy (GUIDE) study, a study that demonstrated that patented crystalline glucosamine sulfate (GS) was superior to placebo and equivalent to acetaminophen in reducing LI after 6 months of treatment.28 However, the present study utilised celecoxib as an active comparator, a NSAID that was recently shown to provide substantially greater clinical effect than acetaminophen in knee OA.12 14
All treatments, including placebo, provided a statistically significant improvement from baseline on pain and function as early as day 30, and this effect persisted until the end of the trial. This is not surprising as a substantial placebo effect was previously reported in trials assessing drugs in OA.29 30 However, both active groups (CS and celecoxib) provided a significantly greater reduction in pain (VAS) and a better improvement in function (LI) than the placebo, after 6 months and 3 months, respectively. With respect to LI, it is interesting to note that celecoxib treatment resulted in a statistically significant change at day 30 compared with placebo, while CS did not. Although impossible to know definitively, this observation may be related to an intrinsic difference in the mechanism of action of the two molecules.
One important consideration in any clinical investigation that uses a pain assessment is how to equate statistical significance with clinical benefit. Indeed, the relevance of statistically significant CS-dependent improvements in OA symptoms in previous trials16–18 has been challenged.21 The EMA 2010 guideline document suggests that the improvement in pain observed with the test article must be clinically relevant, but unfortunately the guideline does not provide an associated numerical threshold. However, a group of European academic scientists and regulators recently published an expert consensus statement in which they suggest that at least a 5 mm difference on a 100 mm VAS between the placebo and active groups constitutes a clinically relevant threshold for a SYSADOAs.22 These recommendations were partially based on the observation that most clinical trials published with SYSADOAs in knee or hip OA show symptomatic improvements in the 5–6 mm range on a 100 mm VAS.31–33 In our study, the difference in pain reduction between CS and placebo is 8.2 mm after 6 months in the ITT analysis. This difference exceeds this range and compares favourably with the previous publications reporting beneficial effects of SYSADOAs in knee or hip OA.31–33
The improvement in pain and function observed in the CS group corresponds to an effect size (ES) of 0.35 for pain and 0.27 for LI, whereas the ES in the celecoxib group was 0.27 for pain and 0.30 for LI. ES ≤0.2 is usually considered as small while ES between 0.2 and 0.5 is defined as medium. An ES value of 0.27 for pain in the celecoxib group is consistent with previous publications8 14 and provides thus external validation of CONCEPT trial data. For CS, an ES of 0.35 for pain is consistent with values previously reported for pharmaceutical-grade GS or CS,8 9 compares well with the reported ES for most NSAIDs8 14 and is two-fold higher than the ES (0.14) commonly reported for acetaminophen in knee OA.8 13
Although both acetaminophen and NSAIDs have been shown to be efficacious in the setting of knee OA, the chronic use of these medicines is known to be associated with frequent and serious adverse events.13 14 It is notable in this regard, that CS, in addition to a robust efficacy profile that is comparable to NSAIDs, also exhibits a safety profile that was similar to placebo in this study and in others.5 8 16 17 21 This combination of therapeutic effect and well-documented safety and tolerability explain why recent guidelines8 9 recommend SYSADOAs, including pharmaceutical-grade CS, as a first-line treatment in the management of knee OA.
In addition to the classic efficacy parameters, pain and LI, regulatory and clinical guidelines continue to place additional emphasis on patient’s perception of their clinical status, thus requiring the use of additional measures to assess treatment outcome. The significantly higher proportion of patients reaching the self-assessed MCII and the significantly greater number of patients ranking their treatment as good or excellent, compared with the placebo group, further reflects the importance of clinical benefits obtained with CS usage.
In conclusion, the CONCEPT study provided evidence that daily administration of 800 mg of 4 &6 CS in patients with symptomatic knee OA lead to improvement in pain and function superior to placebo and similar to the NSAID celecoxib. In addition, we confirmed the excellent safety profile of CS that has been previously observed by others. This compelling benefit-risk profile, in light of the known clinical risks associated with chronic usage of NSAIDs and paracetamol, underscores the potential importance of pharmaceutical-grade CS in the management of knee OA, especially in this older population requiring long-term treatment. More generally, this study corroborates the need for future clinical guidelines on the pharmacological management of knee OA to consider the study design, as well as the composition and quality of the test product, when assessing the effectiveness of SYSADOAs.
Acknowledgments
The authors thank all the investigators of the CONCEPT STUDY: Michael Gengenbacher, Bethesda-Spital, 4052 Basel, Switzerland. Diana P. Frey, Universitatsspital Zurich, Rheumaklinik, Zurich, Switzerland. Marco Matucci Cerinic, Dipartimento di Medicina Interna, Sezione di Reumatologia, Scuola di Spec. In Reumatologia, Unità Operativa di Medicina Interna I e Reumatologia, Azienda Osp. Univ. Di Careggi, Villa Monna Tessa, Firenze, Italy. Radosław Górski, Medica Pro Familia, Warszawa, Poland. Rafał Plebański, Klinika Zdrowej Kości, Łódź, Poland. Jerzy Supronik, Zdrowie Osteo-Medic, Białystok, Poland. Zbigniew Żęgota, SOLB, Ostróda, Poland. Petr Kopsa, Thomayerova nemocnice- Revmatologické a rehabilitačné oddělení, Vídeňská 800, Praha, Czech Republic. Jaroslava Spěváková, Interní a revmatologická ordinace, Břeclav, Czech Republic. Zbyněk Maška, Ortopedická Ambulance, Brno, Czech Republic. Eva Dokoupilová, MEDICAL PLUS, s.r.o., Revmatologická ambulance, Uherské Hradište, Czech Republic. Libor Novosad, Vesalion s.r.o., Ostrava, Czech Republic. The authors thank Giuseppe Mautone and Arturo Lanzarotti, IBSA, Institut Biochimique SA, Via del Piano, 6915 Pambio-Noranco, Switzerland.
References
Supplementary materials
Lay summary
Disclaimer: This is a summary of a scientific article written by a medical professional (“the Original Article”). The Summary is written to assist non medically trained readers to understand general points of the Original Article. It is supplied “as is” without any warranty. You should note that the Original Article (and Summary) may not be fully relevant nor accurate as medical science is constantly changing and errors can occur. It is therefore very important that readers not rely on the content in the Summary and consult their medical professionals for all aspects of their health care and only rely on the Summary if directed to do so by their medical professional. Please view our full Website Terms and Conditions.
Copyright © 2017 BMJ Publishing Group Ltd & European League Against Rheumatism. Medical professionals may print copies for their and their patients and students non commercial use. Other individuals may print a single copy for their personal, non commercial use. For other uses please contact our Rights and Licensing Team.
Footnotes
Contributors All authors agreed to correct the manuscript as per the reviewers recommendations.
Funding The study was sponsored by IBSA Institut Biochimique SA, Pambio- Noranco, Switzerland, a pharmaceutical company marketing Chondroitin Sulfate. The manuscript was entirely written by the first Author (JYR) who received an editorial assistance from IBSA. However, IBSA has no influence on the content of the manuscript. The editorial assistance was limited to the final editing of the manuscript and the submission process through the ARD website.
Competing interests None declared.
Ethics approval Belgium, Italy, Switzerland, Czech Republic, Poland.
Provenance and peer review Not commissioned; externally peer reviewed.