Article Text

Abstract
Objectives Juvenile idiopathic arthritis (JIA) is a heterogeneous group of conditions unified by the presence of chronic childhood arthritis without an identifiable cause. Systemic JIA (sJIA) is a rare form of JIA characterised by systemic inflammation. sJIA is distinguished from other forms of JIA by unique clinical features and treatment responses that are similar to autoinflammatory diseases. However, approximately half of children with sJIA develop destructive, long-standing arthritis that appears similar to other forms of JIA. Using genomic approaches, we sought to gain novel insights into the pathophysiology of sJIA and its relationship with other forms of JIA.
Methods We performed a genome-wide association study of 770 children with sJIA collected in nine countries by the International Childhood Arthritis Genetics Consortium. Single nucleotide polymorphisms were tested for association with sJIA. Weighted genetic risk scores were used to compare the genetic architecture of sJIA with other JIA subtypes.
Results The major histocompatibility complex locus and a locus on chromosome 1 each showed association with sJIA exceeding the threshold for genome-wide significance, while 23 other novel loci were suggestive of association with sJIA. Using a combination of genetic and statistical approaches, we found no evidence of shared genetic architecture between sJIA and other common JIA subtypes.
Conclusions The lack of shared genetic risk factors between sJIA and other JIA subtypes supports the hypothesis that sJIA is a unique disease process and argues for a different classification framework. Research to improve sJIA therapy should target its unique genetics and specific pathophysiological pathways.
- Juvenile Idiopathic Arthritis
- Adult Onset Still's Disease
- Gene Polymorphism
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Video abstract
Introduction
Juvenile idiopathic arthritis (JIA) encompasses a heterogeneous group of chronic childhood arthritides that develop without identifiable cause and last more than 6 weeks.1 ,2 Children with JIA are placed into seven mutually exclusive categories based on clinical presentation: oligoarticular arthritis (oligoJIA) affects four or fewer joints; rheumatoid factor (RF)-negative polyarthritis (RF–polyJIA) involves five or more joints; RF-positive polyarthritis (RF+polyJIA) is analogous to adult rheumatoid arthritis; psoriatic arthritis (PsA) is an arthritis that accompanies psoriasis; enthesitis-related arthritis encompasses non-PsA childhood spondyloarthropathy; systemic arthritis (sJIA, previously known as Still's disease) is characterised by prominent systemic inflammation and has a rare adult-onset counterpart;3 and undifferentiated arthritis includes arthritis that does not fit into any single category.1 ,2
sJIA is among the most severe childhood inflammatory diseases. First described by Sir George Frederic Still over a century ago, sJIA is marked by arthritis and systemic inflammation with quotidian fever, evanescent salmon pink skin rash, lymphadenopathy, hepatosplenomegaly and serositis.2 ,4 It is frequently complicated by macrophage activation syndrome, a potentially lethal form of hemophagocytic lymphohistiocytosis.5 Although sJIA only constitutes approximately 10% of JIA in populations of European descent,1 ,5 its disproportionately large share of the morbidity and mortality observed in JIA6 underscores the importance of understanding and targeting its root causes.
The unique clinical characteristics of sJIA suggest that it is distinct from other forms of JIA, leading to the contention by some that sJIA should be separated from other forms of JIA and labelled as an autoinflammatory disease.7 This has been challenged by identification of autoantibodies in some patients with sJIA.8 Furthermore, while the systemic inflammatory features of sJIA seem to distinguish it from other forms of JIA, most children with sJIA eventually shed these features, leaving up to half of children with a persistent form of arthritis that is similar to the oligoarticular and polyarticular forms of JIA.5 ,9 Finally, significant differential effects of anticytokine agents have been observed between sJIA and other forms of JIA.10 However, due to the highly variable therapeutic responses to each agent in sJIA, this has not concretely advanced our understanding of how sJIA mechanistically relates to other forms of JIA.
One approach to evaluate the similarity of diseases is to examine shared pathophysiology through statistical comparisons of disease-specific genetic association data.11 For example, studies of inflammatory bowel disease and spondyloarthritis have identified shared genetic risk factors, providing rationale for similar treatment choices.11 In JIA, the majority of genetic and genomic investigations have focused on the combination of the most common subtypes, oligoJIA and RF–polyJIA (henceforth referred to in this manuscript as polygoJIA),12 ,13 but until recently,14 because of insufficient numbers of patients with sJIA, there have been only underpowered genetic studies and no genome-wide studies of sJIA. Comparisons of the genomic underpinnings of sJIA relative to other forms of JIA have therefore also been lacking.
To gain insight into the pathogenesis of sJIA, we established the International Childhood Arthritis Genetics (INCHARGE) consortium. Together, we gathered the largest sJIA study population ever assembled, which included 982 children from nine countries on three continents. Using this collection, we performed the first genome-wide association study (GWAS) of sJIA. We recently reported the results of our intensive examination of the major histocompatibility complex (MHC) locus in this study population, which identified the class II human leucocyte antigen (HLA) region as a strong sJIA susceptibility locus.14 Here, we report the findings of the GWAS, beyond the MHC locus. Using the GWAS results, we have performed the first direct comparison of the genetic architecture of sJIA with those of the most common forms of JIA.
Methods
Study design and participants
Peripheral blood specimens were collected from children diagnosed with sJIA according to the International League of Associations for Rheumatology (ILAR) criteria2 by paediatric rheumatologists at participating medical centres in nine countries (see online supplementary text and figure S1). Blood samples were also obtained from geographically matched control subjects. In addition, single nucleotide polymorphism (SNP) genotype data from geographically matched control populations were used, when available. The INCHARGE project was granted institutional review board (IRB) approval by the University of Manchester. Subjects were enrolled in accordance with all local ethics regulations, with the approval of local IRBs at each contributing medical centre, and with informed parental consent.
supplementary data
Genotyping, quality control and imputation
Genomic DNA was extracted from peripheral blood samples. Samples were genotyped at the National Human Genome Research Institute (Bethesda, Maryland, USA) using Human Omni1M arrays (Illumina) in accordance with the manufacturer's protocols. SNP genotype data were stratified by country of origin and rigorous quality control (QC) operations were undertaken separately in each case and control population, as previously reported.14 Principal components analysis and multidimensional scaling were used in each geographically defined case–control collection to generate nine ancestrally matched case–control strata, as previously described.14 Genomic control inflation factors were calculated, per stratum, as an objective metric of ancestral matching.14 An overview of the QC parameters is shown in online supplementary figure S2, and complete details are provided in the online supplementary text and our previous publication.14
SNP genotypes were phased using IMPUTE2,15 and SNP imputation was performed separately for each geographically defined stratum using IMPUTE2 software and the multiancestral 1000 Genomes Project dataset (phase III) as the reference population.16 Genotype probabilities for common markers (case minor allele frequency ≥0.04) that were imputed with high quality (info scores ≥0.8) were included in subsequent analyses.
Statistical analysis
Association testing of genotype probabilities was performed using logistic regression in each geographically defined stratum with SNPTESTv2,15 adjusting for gender and ancestry informative principal components. Association results were meta-analysed using GWAMA.17 Heterogeneity was evaluated in the meta-analyses using the I2 statistic. Weighted genetic risk scores (wGRSs) were calculated and receiver operator characteristic (ROC) curve analyses were performed according to the method of Karlson et al.18 wGRSs were calculated as the sum of the risk allele counts, weighted by the natural logarithm of the OR. The wGRS for polygoJIA (polygo-wGRS) incorporated 23 independent risk alleles reported by Hinks et al12 (see online supplementary table S1). The wGRS for RF+polyJIA (RF+poly-wGRS) was based on the RF+polyJIA-associated wGRS-1119 (see online supplementary table S2). The case and control distributions of risk alleles and wGRSs were evaluated with the Wilcoxon rank-sum test. Association of wGRSs with sJIA was tested by logistic regression, adjusted for ancestry and gender. The ability of wGRSs to discriminate between sJIA and other JIA subtypes was evaluated with ROC curve analysis and calculation of the area under the curve (AUC) using R. Quantile–quantile (Q–Q) plots were generated using the sJIA association data, conditional on sets of polygoJIA-associated SNPs,12 as previously described.20
Results
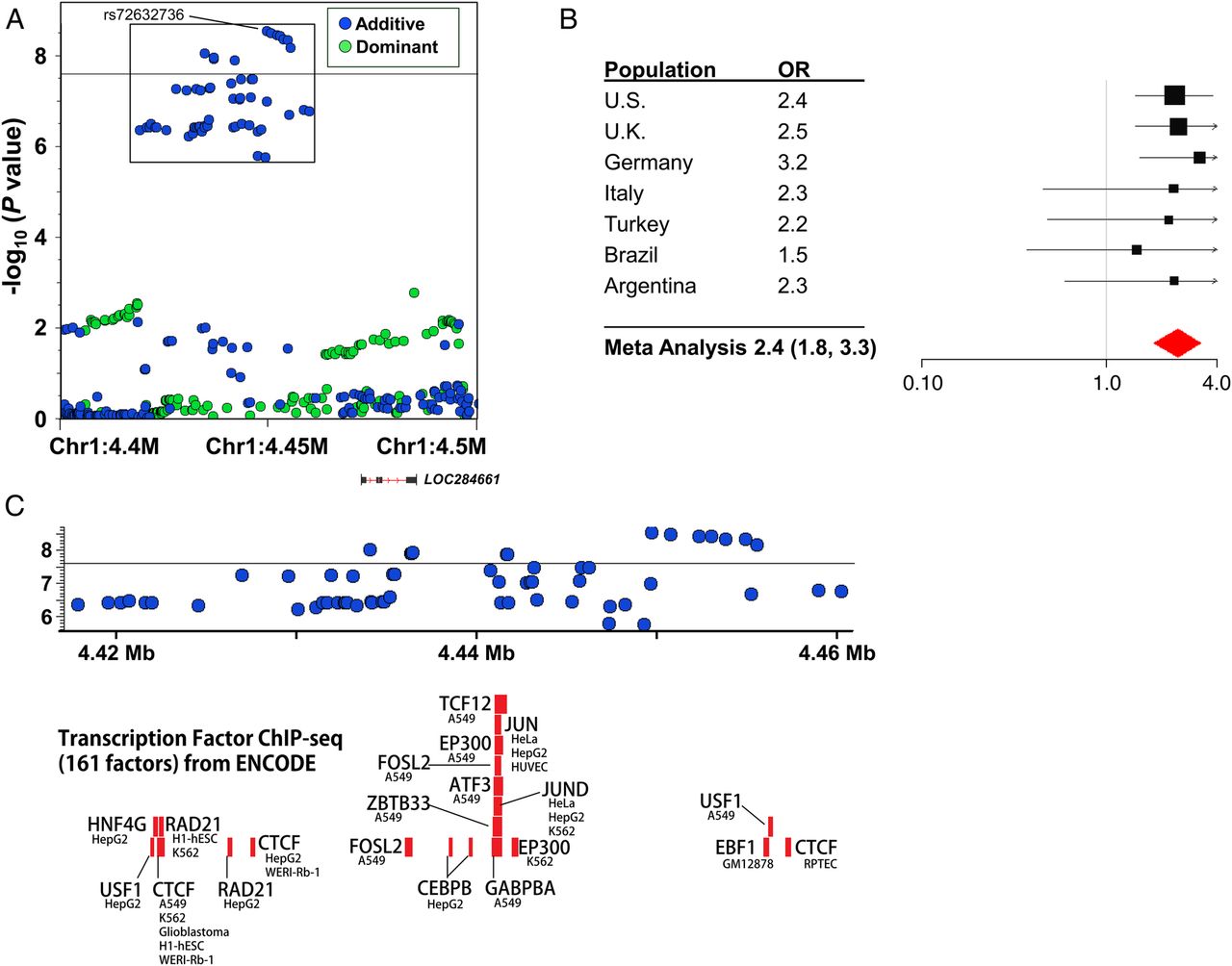
We performed SNP genotyping of 1413 children, including 982 children with sJIA and 431 healthy children. SNP genotype data, in silico, were incorporated from five existing control populations, including 7579 additional subjects, producing a total study population of 8992 individuals. After stringent QC, 770 patients with sJIA and 6947 control subjects were stratified into nine geographically defined and ancestrally matched case–control collections (table 1, see online supplementary text and tables S3 and S4), as previously described.14 Because most in silico control datasets were generated using SNP genotyping platforms different from that used in our study, the final number of SNPs evaluated in strata with in silico data was reduced to the intersection of the different SNP arrays (see online supplementary text and table S4). Imputation produced sets of between 4 147 566 and 6 832 892 imputed SNPs that passed postimputation QC processes (see online supplementary text). Association results were combined by fixed-effect meta-analysis, producing meta-analytic association data for 5 600 610 SNPs (figure 1). This analysis identified two sJIA susceptibility loci with associations exceeding the threshold for genome-wide significance, adjusted for the two models tested (p<2.5×10−8), and 23 loci with highly suggestive evidence of association (p<5×10−6; table 2). With the exception of the MHC locus none of these loci have been previously implicated in sJIA risk or pathophysiology. The strongest sJIA risk locus identified by this study was the MHC locus on chromosome 6 (see online supplementary figure S3). We have recently described this association in great detail in the context of a regional association study of the MHC locus in sJIA.14 Beyond the MHC locus, we identified a novel sJIA susceptibility locus on the short arm of chromosome 1 (1p36.32) whose association also exceeded the threshold for genome-wide significance under the additive model (figures 1 and 2). This locus includes a cluster of 14 sJIA-associated SNPs that span 20.6 kb; the peak SNP is rs72632736 (p=2.9×10−9; OR 2.4 (1.8, 3.3). The association peak is located 20 kb upstream of LOC284661, a long intergenic non-coding RNA, and 263.5 kb upstream of the nearest protein coding gene, AJAP1, encoding adherens junction-associated protein 1. Examination of ENCODE (Encyclopedia of Noncoding DNA Elements) data revealed that the sJIA-associated SNPs overlaid a cluster of transcription factor-binding sites (TFBS) identified by chromatin immunoprecipitation sequencing (ChIP-seq; figure 2) in a variety of cell types; however, none of the top sJIA-associated SNPs were located within the ChIP-seq TFBS.
Summary of SNP datasets from nine sJIA case–control collections after quality control operations
Susceptibility loci with at least suggestive evidence of association with sJIA
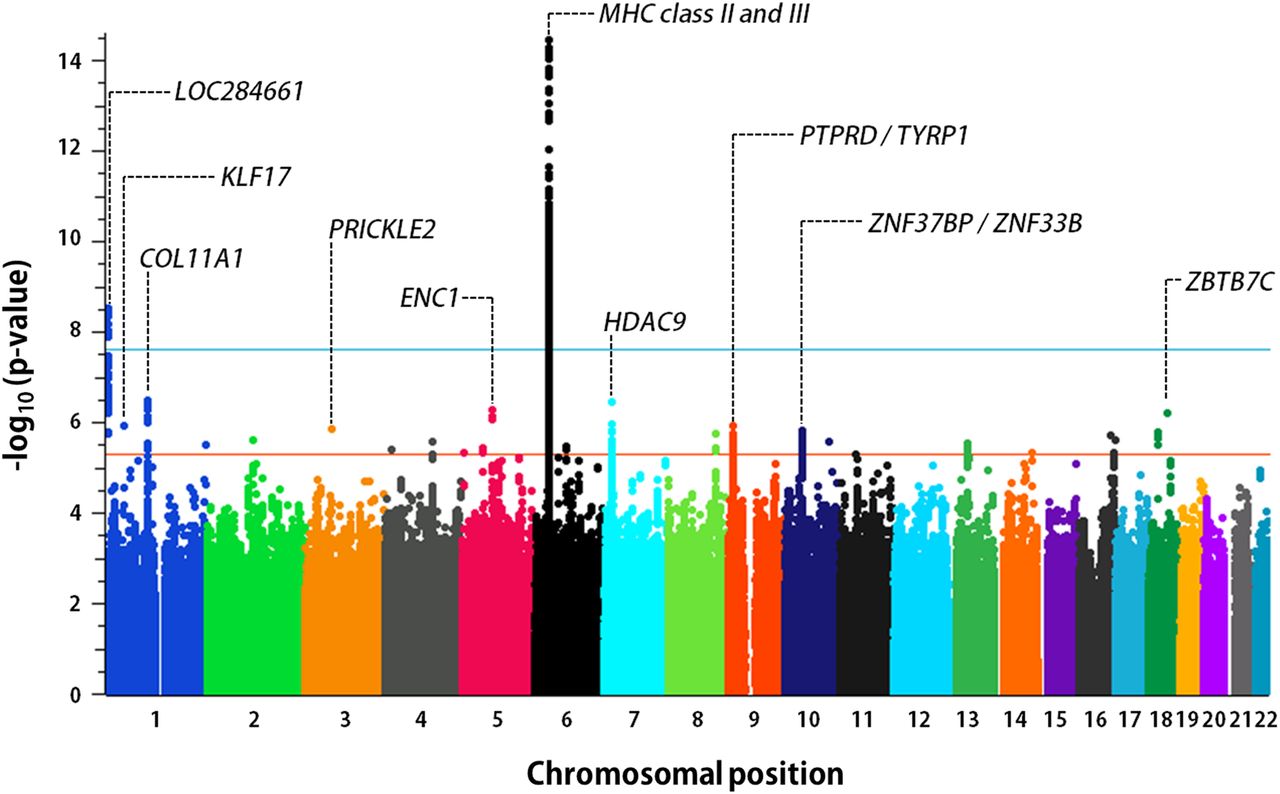
Genome-wide association results from meta-analysis of nine INCHARGE sJIA collections. The threshold of genome-wide significance (p<2.5×10−8) is shown by the blue line, while the orange line marks the level of significance suggestive of association (p<5×10−6). The top 10 sJIA-associated loci are labelled with the name of the nearest gene(s). INCHARGE, International Childhood Arthritis Genetics Consortium; MHC, major histocompatibility complex; sJIA, systemic juvenile idiopathic arthritis.

Systemic juvenile idiopathic arthritis (sJIA) susceptibility locus at chr1p36.32. A regional association plot demonstrates the association between sJIA) and single nucleotide polymorphisms (SNPs) in this region (A). The effect of the peak SNP (rs72632736) in each study population is demonstrated in the forest plot (B). The threshold of genome-wide significance (p<2.5×10−8) is marked by the black horizontal line in (A) and (C). Panel C shows the superimposition of sJIA-associated SNPs (inset box, A) with transcription factor-binding sites determined by chromatin immunoprecipitation (ChIP) sequencing from the Encyclopedia of Noncoding DNA Elements (ENCODE) project.
In addition to the two loci described above, this study identified 23 novel candidate susceptibility loci (figure 1, table 2), two of which are shown in detail in online supplementary figure S4. Importantly, the top 25 sJIA susceptibility loci had scant intersection with the known susceptibility loci of other JIA subtypes. Based on this observation, we sought to compare the genetic architecture of sJIA with those of polygoJIA and RF+polyJIA.
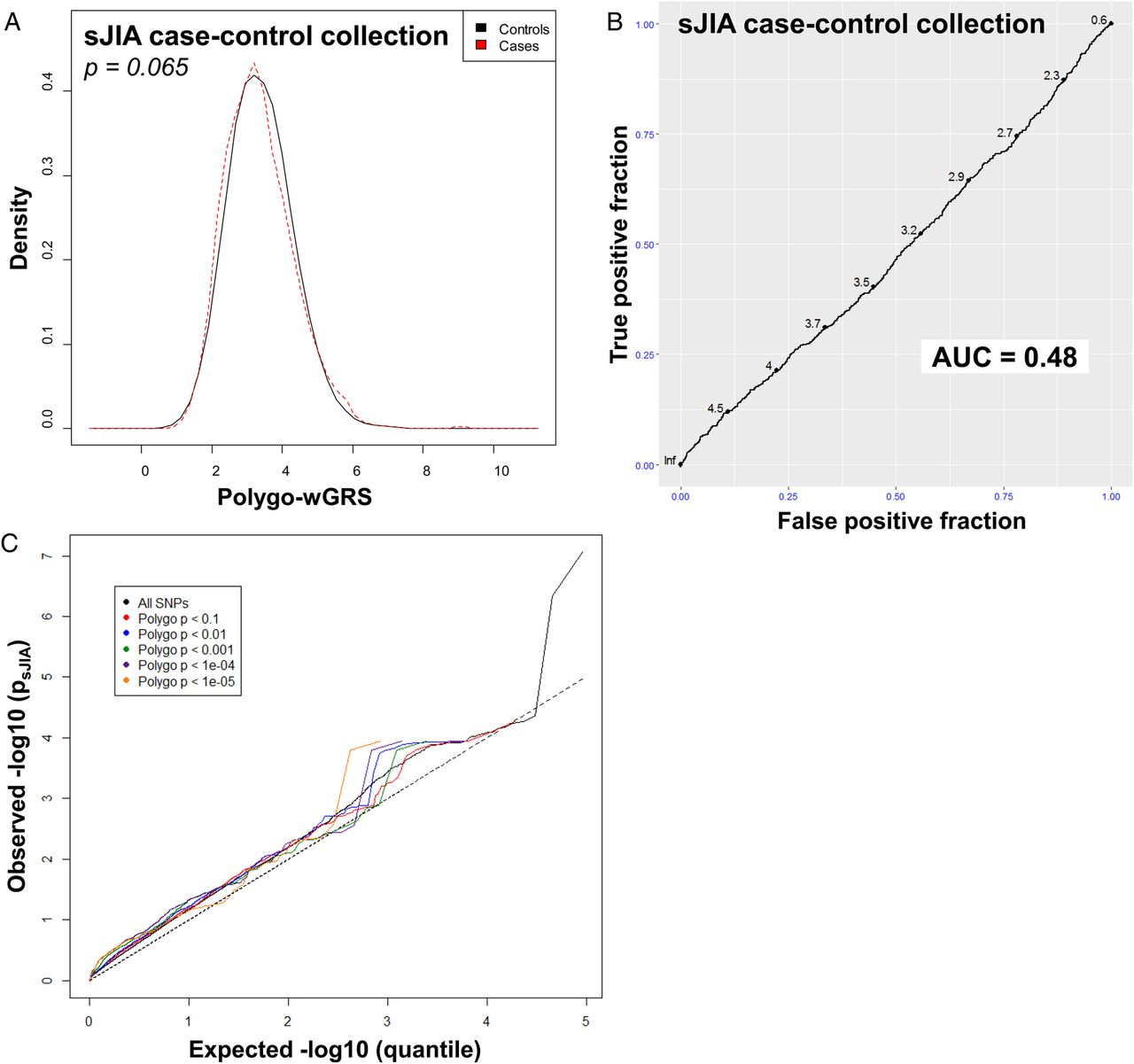
We first examined the 23 polygoJIA-associated loci reported by Hinks et al12 in the sJIA study population and none showed even a modest association with sJIA (see online supplementary tables S5 and S6). To more formally compare sJIA with polygoJIA, we calculated a polygo-wGRS in the sJIA case–control collections based on the same 23 SNPs. The non-parametric Wilcoxon rank-sum test found no difference in the distribution of polygoJIA risk allele counts or polygo-wGRSs between sJIA cases and controls (figure 3, see online supplementary table S7 and figures S5 and S6). Consistent with this, logistic regression analysis found no correlation between the polygo-wGRS and sJIA in any individual stratum or in the full study population (see online supplementary table S7). Analysis of ROC curves in individual strata and the full population found that the AUCs for polygo-wGRS were all close to 0.5, indicating that the polygo-wGRS was no better than random chance at distinguishing sJIA cases from control subjects (figure 3, see online supplementary figure S7). Finally, to expand the scope of our comparison beyond peak SNPs from risk loci, we performed a Q–Q plot-based enrichment analysis to look for shared genetic risk factors between sJIA and polygoJIA (figure 3). By comparing Q–Q plots of polygoJIA-associated SNPs12 at several different significance levels in our sJIA collection, we sought to evaluate pleiotropy in a more global/genomic manner. In the presence of pleiotropy, the slopes of the Q–Q plots of disease A associations are expected to increase as the plotted SNP sets become more strongly associated with disease B, as previously shown.20 In the case of polygoJIA-associated SNPs in sJIA, the slope of the Q–Q plots of sJIA associations did not increase when SNPs of increasingly strong association with polygoJIA were plotted, indicating that there was no enrichment of sJIA-associated variants among polygoJIA-associated variants, and therefore that there was no evidence of pleiotropy (figure 3).
{kind=link}
{kind=link}
{kind=link}
Comparison of the genetic architecture of systemic juvenile idiopathic arthritis (sJIA) with seronegative polyarticular and oligoarticular (polygo) JIA. Kernel density plots display the distribution of polygo-wGRS in sJIA cases and controls from the full study collection (A). p Value was calculated with the Wilcoxon rank-sum test. Receiver operator characteristic (ROC) curves with area under the curve (AUC) calculations demonstrate the performance of polygo-wGRS at predicting sJIA status in the full collection (B). Q–Q plots show the level of association of subsets of polygoJIA-associated single nucleotide polymorphisms in the sJIA population (C).
In addition, we used an RF+poly-wGRS19 to look for shared genetic architecture between sJIA and RF+polyJIA. As was the case with polygoJIA, non-parametric testing revealed no significant difference in the distribution of RF+polyJIA risk alleles (see online supplementary figure S8) or RF+poly-wGRS (see online supplementary figure S9) between sJIA cases and controls in any individual population. Of note, non-parametric testing and logistic regression analysis identified a significant difference in RF+poly-wGRS between sJIA and controls in the full collection (see online supplementary table S8 and figure S10); however, the wGRSs were actually lower in the sJIA cases than in the controls (see online supplementary figure S11). Consistent with these observations, ROC analyses found that the RF+poly-wGRS was not predictive of sJIA (see online supplementary figures S10 and S12). Collectively, these investigations failed to identify any evidence of shared genetic architecture between sJIA and polygoJIA or RF+polyJIA.
Discussion
In this study, two novel susceptibility loci met genome-wide significance criteria for association with sJIA and 23 other loci demonstrated highly suggestive evidence of association. Furthermore, formal comparisons of association data from sJIA with those from polygoJIA and RF+polyJIA have demonstrated that sJIA bears a unique genetic architecture, indicating that its underlying pathophysiological mechanisms are significantly divergent from other forms of JIA. This has important implications and should direct research for future targets of therapeutic intervention for children affected with sJIA.
This is the first large-scale genomic study of sJIA, which includes case–control collections from nine different countries. In a sample of 982 affected children, we identified genome-wide significant evidence of association with SNPs in the class II MHC locus and SNPs on chromosome 1 nearest to an uncharacterised long non-coding RNA gene. This work also identified many additional candidate sJIA susceptibility loci, nearly all of them novel, and aside from the HLA locus, none of these novel loci are associated with any other rheumatic diseases (see online supplementary table S9). The identification of these loci is an important step towards the elucidation of the specific pathways and pathogenic mechanisms in sJIA, which in turn will allow the development of therapies to more specifically target sJIA pathophysiology in affected children. Several of the susceptibility loci that warrant further investigation include strong candidates for therapeutic modulation, and many novel loci or genes that have been poorly studied, to date. Functional investigations are needed to identify and understand the specific mechanisms that underlie the genetic associations.
This study also provided the first opportunity to demonstrate that sJIA did not share heritable risk factors with the more common oligoarticular and polyarticular forms of JIA. There was no intersection of the top susceptibility loci of sJIA with those of polygoJIA or RF+polyJIA. Even within the class II MHC region, which harbours disease-associated genetic variation in each of these categories of JIA, the subtype-specific risk factors (SNPs, HLA alleles and HLA haplotypes) are not shared between subtypes. Using a combination of genetic risk scores and enrichment analysis, this study reveals an absence of shared genetic architecture between sJIA and either polygoJIA or RF+polyJIA, despite often sharing a chronic arthritis feature with polygo or RF+polyJIA. It could be that as a clinical feature, arthritis is a non-specific finding that is present in many different conditions, including infections, malignancies, autoimmune disorders and autoinflammatory conditions. These distinct genetic data provide hard evidence that these conditions differ in pathophysiology, strongly supporting the clinical distinction between sJIA and the other JIA subtypes. Considering the ongoing discussions about restructuring the JIA nomenclature, these studies will help inform and guide the debate surrounding sJIA7 and how it should be classified.
The genetic dissimilarity of sJIA and other JIA subtypes has important therapeutic implications for children with sJIA. Currently, the treatment of sJIA presents physicians with a clinical conundrum, with no single, universally effective therapeutic approach. Prior to the era of biological response-modifying agents, sJIA was treated with disease-modifying antirheumatic drugs, including methotrexate, with a rationale for use extrapolated from other forms of JIA; there were no clinical trials and only limited outcome studies describing their effectiveness in sJIA.10 In the absence of clear therapeutic alternatives, and despite the limited evidence of efficacy, methotrexate remains an accepted therapeutic option in the consensus treatment protocol.21 Similarly, therapies targeting the cytokine tumour necrosis factor-α are highly effective in the treatment of other forms of JIA,22 but show only modest effect in children with sJIA.10 Today, even with the most effective treatments for sJIA directed against the inflammatory cytokines interleukin (IL)-1 and IL-6,10 a sizable proportion of children continue to have active disease, with chronic arthritis persisting in nearly 40% of children in a recent study.9 Currently, the only widely effective treatment for sJIA remains large doses of glucorticoids.10 There is clearly an imperative to look for root causes of sJIA to identify better targets for therapy and prevent the development of persistent, disabling arthritis.
Although it is necessary to better understand the function of the risk alleles identified by this study, the results may identify genetic profiles that can be used to determine appropriate therapeutic interventions. To this point, two susceptibility loci are of particular therapeutic interest in sJIA: the class II HLA locus and HDAC9, encoding histone deacetylase 9. Given that class II HLA molecules present peptide antigens to T-cell receptors on CD4+ T cells, resulting in their activation, one may predict that therapeutic modulation of T-cell activation would be an effective strategy in the treatment of sJIA. In fact, abatacept, which reduces T-cell activation through costimulatory inhibition, has shown promising results in children with the chronic, persistent arthritis of sJIA23 ,24—a subset of patients with sJIA who are particularly refractory to therapeutic intervention.5 Based on these observations, it may be reasonable to use abatacept in children with sJIA. HDAC9 confers important epigenetic effects through deacetylation of histone proteins, while also regulating critical innate immune processes, including Toll-like receptor signalling and the development of regulatory T cells, via deacetylation of non-histone targets.25–28 Despite the fact that HDAC9 was only suggestively associated with sJIA, a pilot study of the non-specific HDAC inhibitor, gavinostat, produced promising preliminary results in children with sJIA,29 raising the possibility that HDAC inhibition represents another plausible targeted therapeutic strategy in sJIA.
At a time when an emphasis is being placed on the personalisation of medicine, it is important that we move away from broad classifications based on non-specific clinical observations and move towards the use of molecular and genetic data in establishing diagnoses, as well as pathophysiology. In turn, clinical practice will advance as these data are translated into targeted therapeutic approaches. Perhaps it is time to separate this condition from JIA all together to make clear that it is fundamentally different from any other form of JIA and needs to be considered and treated differently. Given that the currently available treatments for this condition are still imperfect, it remains imperative to continue to employ contemporary investigative approaches in sJIA, to elucidate its pathophysiology and to identify the next generation of therapeutic strategies.
References
Footnotes
Handling editor Tore K Kvien
Twitter Follow NIAMS/NIH/DHHS @NIH_NIAMS; Ricardo Russo @el_reumatologo; University of Manchester, Centre for Musculoskeletal Disease @UoMMskResearch
Collaborators Full membership of collaborating consortia are listed in the supplementary text: British Society of Pediatric and Adolescent Rheumatology Study Group, Inception Cohort of Newly Diagnosed Patients with Juvenile Idiopathic Arthritis Study Group, Childhood Arthritis Prospective Study Group, Randomized Placebo Phase Study of Rilonacept in sJIA Investigators, Sparks-Childhood Arthritis Response to Medication Study Group and Biologically Based Outcome Predictors in JIA Group.
Contributors All authors participated in study design; AAG, DF, AM, MG, SÖ, SP, ASZ, JFB, NTI, EDM, RR, CL, MOEH, SO, RSMY, AMR, LRW, JA, J-PH, AR-W, KM, KT, ED, BSPAR, ICON-JIA, CAPS, RAPPORT, CHARMS, BBOP, RHD, JPA, MIK, KMK, LCK, DP, SWS, MEA-R, ED, XE and AG provided samples for the study; MJO, VLA, EFR, AH, IT, EZ, PW and WT performed the research and analysis and interpreted the data; all authors drafted and/or substantively edited the manuscript and have thoroughly reviewed and approved of the content.
Funding This work was supported by the Intramural Research Programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases (Z01-AR041198 to MJO) and the National Human Genome Research Institute (Z01-HG200370 to DLK) of the National Institutes of Health (NIH). Additional funding was provided by NIH grants R01-AR059049 (AAG), R01AR061297 (EDM), R01-AR060893 (SP), P30-AR47363 and P01-AR48929 (ST), AG030653, AG041718 and AG005133 (MIK) and U01-DK062420 and R01-DK076025 (RHD); Arthritis Research UK Grant 20385 (WT); the German Federal Ministry of Education and Research (BMBF project 01ER0813) for the ‘ICON-JIA’ inception cohort (KM and DF); the Val A. Browning Charitable Foundation (JFB); the Marcus Foundation (SP); the Proyecto de Excelencia (CTS-2548) of the Andalousian Government (MA-R) and the Swedish Association Against Rheumatism (MA-R). IT and EZ were supported by the Wellcome Trust (098051). WT and JC are funded by the National Institute for Health Research Biomedical Research Unit Funding Scheme. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. The CAPS study was funded by Arthritis Research UK Grant 20542. WT, AH, and JC are supported by the Manchester Academic Health Sciences Centre (MAHSC). SPARKS-CHARMS was funded by grants from SPARKS UK (08ICH09 and 12ICH08), the Medical Research Council (MR/M004600/1) and the UK National Institute for Health Research GOSH Biomedical Research Centre. The BBOP study was supported by the Canadian Institutes of Health Research and the Arthritis Society (CIHR funding reference number 82517) and the Canadian Arthritis Network (funding reference SRI-IJD-01). This research was supported in part by the Cincinnati Children's Research Foundation and its Cincinnati Genomic Control Cohort. The authors acknowledge the use of DNA from the UK Blood Services collection of Common Controls (UKBS-CC collection), which is funded by Wellcome Trust grant 076113/C/04/Z and by the USA NIH research programme grant to the National Health Service Blood and Transplant (RP-PG-0310-1002). The authors acknowledge the use of DNA from the British 1958 Birth Cohort collection, which is funded by the UK Medical Research Council grant G0000934 and the Wellcome Trust grant 068545/Z/02. This study used the computational resources of the Biowulf system at the NIH, Bethesda, MD (http://biowulf.nih.gov).
Competing interests AAG: consulting fees and honoraria from Novimmune, Novartis, Roche. AM: consulting fees and honoraria from Abbvie, Boehringer, Celgene, CrescendoBio, Janssen, Meddimune, Novaris, NovoNordisk, Pfizer, Sanofi Aventis, Vertex and Servier, contributions have been received by G. Gaslini Hospital and the PRINTO network by BMS, GlaxoSmithKline, Hoffman-La Roche, Novartis, Pfizer, Sanofi Aventis, Schwarz Biosciences, Abbot, Francesco Angelini S.P.A., Sobi, and Merck Serono. MG: consulting and speaker fees from SOBI and Novartis, unrestricted grants to Eurofever from SOBI and Novartis. SP: consulting fees from Novartis. EDM: consulting fees from Novartis. LRW: speaker fees from Pfizer.
Ethics approval University of Manchester.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The quality control processed directly genotyped SNP genotype data from sJIA cases genotyped for this study will be deposited into the National Institutes of Health’s Database of Genotypes and Phenotypes, where allowable by the Ethics and consent documents. The future use of these data will be dictated by the terms of Ethics and consent documents and the institutional certifications provided by the collaborating centres.