Article Text

Abstract
Objectives To compare efficacy and safety of sarilumab monotherapy with adalimumab monotherapy in patients with active rheumatoid arthritis (RA) who should not continue treatment with methotrexate (MTX) due to intolerance or inadequate response.
Methods MONARCH was a randomised, active-controlled, double-blind, double-dummy, phase III superiority trial. Patients received sarilumab (200 mg every 2 weeks (q2w)) or adalimumab (40 mg q2w) monotherapy for 24 weeks. The primary end point was change from baseline in 28-joint disease activity score using erythrocyte sedimentation rate (DAS28-ESR) at week 24.
Results Sarilumab was superior to adalimumab in the primary end point of change from baseline in DAS28-ESR (−3.28 vs −2.20; p<0.0001). Sarilumab-treated patients achieved significantly higher American College of Rheumatology 20/50/70 response rates (sarilumab: 71.7%/45.7%/23.4%; adalimumab: 58.4%/29.7%/11.9%; all p≤0.0074) and had significantly greater improvement in Health Assessment Questionnaire-Disability Index (p=0.0037). Importantly, at week 24, more patients receiving sarilumab compared with adalimumab achieved Clinical Disease Activity Index remission (7.1% vs 2.7%; nominal p=0.0468) and low disease activity (41.8% vs 24.9%; nominal p=0.0005, supplemental analysis). Adverse events occurred in 63.6% (adalimumab) and 64.1% (sarilumab) of patients, the most common being neutropenia and injection site reactions (sarilumab) and headache and worsening RA (adalimumab). Incidences of infections (sarilumab: 28.8%; adalimumab: 27.7%) and serious infections (1.1%, both groups) were similar, despite neutropenia differences.
Conclusions Sarilumab monotherapy demonstrated superiority to adalimumab monotherapy by improving the signs and symptoms and physical functions in patients with RA who were unable to continue MTX treatment. The safety profiles of both therapies were consistent with anticipated class effects.
Trial registration number NCT02332590.
- Rheumatoid Arthritis
- Treatment
- DMARDs (biologic)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Biological disease-modifying antirheumatic drugs (bDMARDs) targeting inflammatory cytokines, such as tumour necrosis factor α (TNF-α) or interleukin 6 (IL-6) via the IL-6 receptor (IL-6R), have expanded the treatment options for patients with rheumatoid arthritis (RA).1–3 Emerging data have demonstrated that patients with inadequate response to conventional synthetic DMARDs (csDMARDs; eg, methotrexate (MTX)) benefit from early and intensive therapy with the addition of bDMARDS, resulting in better preservation of joint structure and function.4–9 Yet, nearly one-third of patients with RA use biologics as monotherapy due to MTX intolerance or contraindication.10–13 In addition, increasing data from real-world clinical practice and prescription drug registries across multiple countries indicate that bDMARDs are frequently used as monotherapy, either at the discretion of the physician or because of patient preference.13–17 The widespread use of bDMARD monotherapy calls for more comparative data to support the optimal selection of approved bDMARDs in clinical practice.
Therapeutic targeting of the IL-6R has been a major advance in the effective treatment of RA, as IL-6R plays a key role in mediating the underlying disease pathophysiology and clinical manifestations of RA.18–22 In patients with RA, elevated levels of IL-6 in the serum and synovial fluid tightly associate with synovitis, systemic inflammation, bone metabolism, fatigue and joint destruction.23
Sarilumab is a human IgG1 monoclonal antibody that binds specifically to both soluble and membrane-bound IL-6Rs (sIL-6Rα and mIL-6Rα) and has been shown to inhibit IL-6-mediated signalling through these receptors. In two previous phase III trials, sarilumab administered subcutaneously at 150 and 200 mg every 2 weeks (q2w) was effective in several patient populations with RA, including MTX inadequate responders24 and those with an inadequate response or intolerance to TNF inhibitors.25 In MTX inadequate responders, the addition of sarilumab inhibited radiographic progression and, in both studies, sarilumab achieved rapid and sustained improvement in disease activity and improved physical function with a manageable safety and tolerability profile consistent with IL-6R blockade.24–27
Adalimumab is a globally approved bDMARD targeting TNF-α that is recommended for use in patients who fail to achieve clinical remission with csDMARDs (including MTX) and is an approved monotherapy for those unable to take csDMARDs because of intolerance or contraindication.2 ,28 The objective of the phase III MONARCH trial (NCT02332590) was to compare the efficacy and safety of sarilumab and adalimumab monotherapy in patients with active RA who were unsuitable candidates for continued treatment with MTX due to intolerance or inadequate response. Results from this study address the need for data comparing biological monotherapy performance, to help better define strategies for the choice and optimal sequencing of available therapeutics suited for real-world clinical practice.
Methods
Study design
MONARCH was a multicentre, randomised, active-controlled, double-blind, double-dummy, phase III superiority trial conducted in 86 study centres in Europe, Israel, Russia, South Africa, South America, South Korea and the USA. The first patient was enrolled on 11 February 2015 and the last patient completed week 24 on 20 January 2016. After 24 weeks, patients had the option to enrol in an open-label extension. Results from the 24-week, double-blind treatment period are presented.
Patients were centrally randomised using an interactive voice response system to receive sarilumab 200 mg q2w plus placebo q2w (n=184) or adalimumab 40 mg q2w plus placebo (n=185) in prefilled matching syringes for subcutaneous administration for 24 weeks. Treatment and matching placebo were provided in kits suitable for double-dummy blinding; investigators did not have access to randomisation information except under exceptional medical circumstances. After week 16, dose escalation to weekly administration of adalimumab or matching placebo in the sarilumab group was permitted for patients who did not achieve ≥20% improvement in tender and swollen joint counts.
The protocol was approved by the appropriate ethics committees/institutional review boards and each patient gave written consent before participation in the study. The study was conducted in compliance with institutional review board regulations, the International Conference on Harmonization Guidelines for Good Clinical Practice and the Declaration of Helsinki.
Patient population
Eligible patients were ≥18 years at baseline and those who fulfilled the 2010 American College of Rheumatology (ACR)/European League Against Rheumatism Classification Criteria for RA29 and ACR class I–III functional status, based on the 1991 revised criteria.30 Patients were included if they had active RA, defined as ≥6 of 66 swollen and ≥8 of 68 tender joints and high-sensitivity C reactive protein (CRP) ≥8 mg/L or erythrocyte sedimentation rate (ESR) ≥28 mm/hours and 28-joint disease activity score using ESR (DAS28-ESR) >5.1 assessed between screening and randomisation, with disease duration ≥3 months and were, per investigator judgement, either intolerant of or considered inappropriate candidates for continued treatment with MTX, or inadequate responders if treated with an adequate MTX dose (10–25 mg/week or 6–25 mg/week for patients within Asia-Pacific region) for ≥12 weeks. Patients with prior bDMARD experience were excluded.
Efficacy end points
The primary efficacy end point was change from baseline in DAS28-ESR at week 24. Secondary efficacy end points at week 24 included DAS28-ESR remission (<2.6); the Health Assessment Questionnaire-Disability Index (HAQ-DI); ACR 20% (ACR20), 50% (ACR50) and 70% (ACR70) responses; Medical Outcomes Short Form 36 Health Survey (V.2) (SF-36) physical component summary (PCS) score and mental component summary (MCS) score and Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F). Clinical Disease Activity Index (CDAI), a pre-specified secondary end point, was not part of the hierarchy as it was not consistent with regulatory guidance. For a list of end points, see online supplementary table S1.
supplementary table 1
Safety
Safety assessments included incidence of treatment-emergent adverse events (AEs), serious AEs (SAEs) reported by investigators, along with measured laboratory tests. AEs were described in the Medical Dictionary for Regulatory Activities (V.18.1) preferred-term level, whereas AEs of special interests were identified using pre-specified search criteria. Antidrug antibody (ADA) positivity at two or more consecutive samplings or the last sample analysed during the AE period was classified as persistent.
Statistical analysis
A sample size of 170 patients per group was needed to provide at least 90% power to demonstrate that sarilumab was superior to adalimumab by at least 0.6 units (a clinically relevant difference26) on the DAS28-ESR scale using a SD of 1.7 based on prior trials.26 Efficacy analyses were conducted in the intent-to-treat (ITT) population, which included all randomised patients, including those who increased the dose frequency of adalimumab or matching placebo. Data collected after permanent treatment discontinuation were excluded. Sensitivity analyses and statistical methods are described in the online supplementary appendix.
supplementary appendix
Results
Patient demographics, baseline characteristics and disposition
The ITT population consisted of 369 patients (185 in the adalimumab group and 184 in the sarilumab group; figure 1). Baseline characteristics and treatment history were generally balanced between groups (table 1).
Patient demographics and disease characteristics

Flow diagram showing patient disposition. *Primary reasons for patient ineligibility were meeting the exclusion criteria related to tuberculosis (12.0%) or failure to meet the inclusion criterion for severity of disease (8.1%). †One patient was randomised but not treated in the adalimumab group. ‡The actual number of patients who received a dose-escalation kit on the basis of meeting protocol criteria were 6 (3.2%) in the adalimumab group and 5 (2.7%) in the sarilumab group. q2w, every 2 weeks.
Patients in the sarilumab group tended to have lower baseline CRP and longer RA duration compared with patients in the adalimumab group, although DAS28-CRP and DAS28-ESR were comparable between groups. Percentages of MTX non-responders and MTX-intolerant patients were also balanced (table 1). The mean highest weekly prior MTX dose was 16.9 mg/week.
The treatment period was completed by most patients (sarilumab: 90%, adalimumab: 84%), with AEs the most common cause of discontinuation (figure 1). The safety population consisted of all patients who received at least one dose of study medication; this population included 368 patients because one patient was randomised to the adalimumab group in error and did not receive study medication.
Efficacy
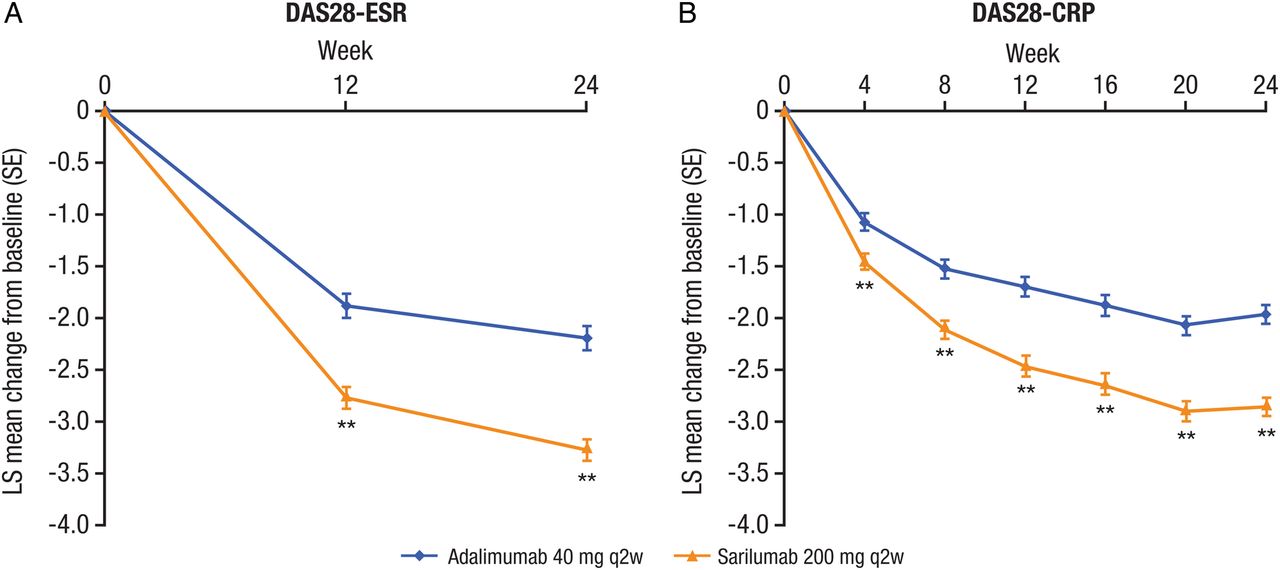
The primary end point of the study was achieved: sarilumab 200 mg q2w was superior to adalimumab 40 mg q2w in mean change from baseline to week 24 in DAS28-ESR (−3.28 vs −2.20; difference: −1.08; 95% CI −1.36 to −0.79; p<0.0001) (table 2; figure 2A).
Hierarchical order of primary and secondary end points at week 24

Change from baseline in (A) DAS28-ESR and (B) DAS28-CRP in patients receiving adalimumab 40 mg q2w or sarilumab 200 mg q2w. **p<0.001 versus adalimumab (DAS28-CRP are nominal p values). CRP, C reactive protein; DAS28-ESR, 28-joint disease activity score using erythrocyte sedimentation rate; LS, least squares; q2w, every 2 weeks.
Improvements in DAS28-ESR were greater by week 12 in the sarilumab group compared with the adalimumab group (−2.77 vs −1.88; difference: −0.89; 95% CI −1.18 to −0.59; nominal p<0.0001). Compared with adalimumab, the odds of achieving DAS28-ESR remission with sarilumab were approximately three times greater at week 12 (OR: 2.61; 95% CI 1.31 to 5.20; nominal p=0.0051) and approximately five times greater at week 24 (OR: 4.88; 95% CI 2.54 to 9.39; p<0.0001) (figure 3A).

{kind=link}
{kind=link}
{kind=link}
Incidence of (A) DAS28-ESR remission or LDA, (B) ACR20, ACR50 and ACR70 response from weeks 4 to 24, (C) CDAI remission or LDA and (D) HAQ-DI responders achieving ≥0.22 or ≥0.3 units of improvement in patients receiving adalimumab 40 mg q2w or sarilumab 200 mg q2w. *p<0.05 versus adalimumab; **p<0.01 versus adalimumab (CDAI and HAQ-DI responders at week 24 are nominal p values); †p<0.0001 versus adalimumab. ACR, American College of Rheumatology; CDAI, Clinical Disease Activity Index; DAS28-ESR, 28-joint disease activity score using erythrocyte sedimentation rate; HAQ-DI, Health Assessment Questionnaire-Disability Index; LDA, low disease activity; q2w, every 2 weeks.
Sensitivity analyses (see online supplementary appendix) for the primary endpoint were consistent with the primary analysis (see online supplementary table S2). Additionally, in a pre-specified subgroup analysis, sarilumab demonstrated greater change from baseline in DAS28-ESR at week 24 compared with adalimumab, regardless of previous MTX response (treatment-by-subgroup interaction: intolerant vs inadequate response, p=0.2163; see online supplementary table S3).
supplementary table 2
supplementary table 3
Change in DAS28-CRP at week 24 was consistent with DAS28-ESR (−2.86 vs −1.97; difference: −0.88; 95% CI −1.14 to −0.63; nominal p<0.0001). At the first assessment (week 4), mean change in DAS28-CRP was larger in the sarilumab group compared with the adalimumab group (−1.46 vs −1.08; difference: −0.38; 95% CI −0.59 to −0.16; nominal p=0.0005) and the numerical difference between groups continued to increase throughout the study (figure 2B; see online supplementary table S4).
supplementary table 4
Sarilumab also demonstrated greater efficacy compared with adalimumab in CDAI, a measure of clinical response independent of acute-phase reactants that may favour IL-6 inhibition. Patients receiving sarilumab had a lower mean CDAI score at weeks 12 and 24 compared with patients taking adalimumab (week 24: −28.9 vs −25.2; difference: −3.74; 95% CI −6.02 to −1.47; nominal p=0.0013; see online supplementary table S4). At week 24, more patients receiving sarilumab achieved CDAI remission (13/184 (7.1%) vs 5/185 (2.7%); nominal p=0.0468) and CDAI low disease activity (77/184 (41.8%) vs 46/185 (24.9%); post hoc nominal p=0.0005) compared with those receiving adalimumab (figure 3B; see online supplementary table S4).
The proportion of patients who achieved an ACR20/50/70 response at week 24 was significantly greater in the sarilumab group (71.7%/45.7%/23.4%) than the adalimumab group (58.4%/29.7%/11.9%; all p≤0.0074), with differences observed as early as week 8 (figure 3C). In all three response categories, the between-group difference was >10%. At week 24, both sarilumab and adalimumab had greatly reduced the mean tender (9.0 vs 9.9, out of 68 assessed; p=0.0986) and swollen (4.2 vs 4.8, out of 66 assessed; p=0.0446) joint counts (ACR components described in online supplementary table S4).
The mean improvement in HAQ-DI score from baseline to week 24 was significantly greater in the sarilumab group compared with the adalimumab group (−0.61 vs −0.43; difference: −0.18; 95% CI −0.31 to −0.06; p=0.0037) (table 2). The proportion of patients who demonstrated a clinically meaningful improvement of ≥0.22 units as well as the more stringent ≥0.3 units was higher for patients receiving sarilumab versus those receiving adalimumab (nominal p<0.01 for both) (figure 3D).
At week 24, sarilumab-treated patients had significantly greater improvement in the SF-36 PCS compared with adalimumab-treated patients and improvements were observed as early as week 12 (table 2). Both groups demonstrated similar improvement in SF-36 MCS at week 24. An improvement from baseline to week 24 in FACIT-F score was observed in both groups, with a trend towards greater improvement in the sarilumab group (table 2).
Safety
The incidence of AEs (∼64%, both groups) and SAEs (adalimumab, 12 (6.5%) vs sarilumab, 9 (4.9%)) and the rate of discontinuations (adalimumab, 13 (7.1%) vs sarilumab, 11 (6.0%)) were similar between groups (table 3).
Safety results
One patient in the sarilumab group died of acute cardiac failure secondary to aortic dissection and papillary muscle rupture on day 36.
The incidence of infections was similar between groups (adalimumab, 27.7%; sarilumab, 28.8%). Two patients in each treatment group experienced a serious infection: one mastitis and one infective bursitis with sarilumab and one bacterial arthritis and one upper respiratory tract infection with adalimumab (table 3).
One patient in the adalimumab group developed multiple sclerosis. One patient in the sarilumab group was diagnosed with demyelinating polyneuropathy; symptoms began before randomisation. No cases of gastrointestinal perforation, anaphylaxis or lupus-like syndrome were reported in either group.
Injection site reactions were reported in 8 patients (4.3%) in the adalimumab group and 17 patients (9.2%) in the sarilumab group; two patients in the sarilumab group discontinued as a result. In both groups, the reactions were mild to moderate and the most common AE was erythema.
Neutrophil counts <1.0 G/L occurred more frequently in the sarilumab group compared with the adalimumab group (see online supplementary table S5). Sixteen patients (8.7%) receiving sarilumab and two patients (1.1%) receiving adalimumab had an absolute neutrophil count (ANC) between ≥0.5 and 1 G/L and three patients (1.6%) receiving sarilumab reported an ANC of <0.5 G/L. There was no evidence of an association between decreases in neutrophil counts and risk of infections or serious infections. Infection rates (adalimumab, 51 (27.7%); sarilumab, 53 (28.8%)) were similar between both groups, despite differences in incidence of neutropenia (table 3).
supplementary table 5
The incidence of alanine aminotransferase (ALT) increases between 1 and 3×upper limit of normal (ULN) was 33.7% in the sarilumab group versus 21.2% in the adalimumab group (see online supplementary table S5). ALT elevations >5×ULN were similar between groups (sarilumab, 1 (0.5%) vs adalimumab, 2 (1.1%)). The mean increase in ALT at week 24 was greater in the sarilumab group (6.1 IU/L) compared with the adalimumab group (2.1 IU/L).
Reported AEs of serum lipid elevations occurred more frequently in the adalimumab group (8 (4.3%)) than in the sarilumab group (3 (1.6%)) and five patients in the adalimumab group versus two patients in the sarilumab group initiated a lipid-modifying agent during the treatment period. While patients in the sarilumab group demonstrated a greater mean increase from baseline in low-density lipoprotein (LDL) cholesterol compared with patients in the adalimumab group (0.27 mmol/L vs no change), the majority of sarilumab-treated patients did not shift upward in LDL classification (see online supplementary table S6).
supplementary table 6
ADAs were measured in the sarilumab group; 13 patients tested positive during the AE period. Of these, 5 (2.7%) were defined as persistent ADA because the last sample measured was positive in the ADA assay. No neutralising ADA was detected. The presence of ADA was not associated with hypersensitivity reactions or discontinuations due to lack or loss of efficacy (see online supplementary table S7).
supplementary table 7
Discussion
Use of biologics as monotherapy is an important therapeutic option for patients with RA when use in combination with MTX or other csDMARDs is unsuitable.13 In MONARCH, sarilumab was superior to adalimumab in the reduction of disease activity and improvement in the signs and symptoms of RA, as demonstrated by greater reduction in DAS28-ESR. Greater efficacy with sarilumab versus adalimumab was also observed with CDAI, illustrating that the benefits of sarilumab monotherapy extend beyond the pharmacodynamic effects on acute-phase reactants. The odds of CDAI and DAS28 disease remission were greater with sarilumab compared with adalimumab, despite the allowance of adalimumab dose escalation. Additionally, there was no difference in the magnitude of response for patient populations intolerant to MTX versus those with inadequate response, indicating that the robust efficacy outcomes observed with sarilumab were independent of prior MTX use or response.
From the patient's perspective, the most important benefits of RA treatment are to improve functional disability, pain and fatigue.31–33 Relative to adalimumab, patients receiving sarilumab reported greater improvement in their health status as reflected by differences in SF-36 PCS, HAQ-DI and pain visual analogue scale scores, along with a trend towards greater improvement in fatigue. While the numerical reductions in tender and swollen joint counts were similar between treatment groups, sarilumab-treated patients had less pain and showed greater improvement in global assessments (see online supplementary table S4). These differences reflect that the superiority of objective clinical outcomes observed with sarilumab treatment translate into patient benefits as assessed across a range of patient-reported outcomes.
The safety profiles of sarilumab and adalimumab monotherapy observed in MONARCH were generally comparable. Numerically more patients discontinued with adalimumab because of AEs compared with the sarilumab group. The most common AEs associated with sarilumab were neutropenia and injection site erythema (mostly mild to moderate), while headache and exacerbations of RA were more common in the adalimumab group. Though neutropenia was more common in the sarilumab group, infection rates were similar between study arms. ADA was monitored in the sarilumab group. There was no relationship between ADA and discontinuations due to lack of efficacy or with hypersensitivity reactions and all instances were non-neutralising.
Overall, the safety and tolerability of sarilumab is consistent across studies24 ,25 and comparable with therapeutic targeting of the IL-6 pathway.26 ,27 In MONARCH, changes in laboratory values in the sarilumab group, including neutropenia, liver transaminases and total cholesterol, were expected class effects. While the mechanism of neutropenia remains unclear, studies have shown that blockade of IL-6R does not affect neutrophil function.34 This is consistent with MONARCH and previous sarilumab studies,24 ,25 demonstrating that decreased neutrophil counts were not associated with a concurrent increase in infection rate.
In MONARCH, sarilumab monotherapy was associated with lower incidence of ALT elevations compared with previous studies in which sarilumab was administered in combination with csDMARDs.24 ,25 Because IL-6 aids in protecting the liver from hepatotoxic agents,35 IL-6 blockade in combination with MTX, a known hepatotoxicant, may exacerbate the MTX hepatotoxicity observed in some patients.36 ,37
Head-to-head trials comparing the efficacy and safety of two different bDMARD monotherapies in a clinically relevant patient population can aid in defining strategies for optimal patient care. MONARCH expands on results from ADACTA (NCT01119859),26 showing that blockade of IL-6R is effective in MTX-intolerant patients and in patients with inadequate response to MTX, demonstrating that MTX history does not impact response to therapy. While both MONARCH and ADACTA showed superior efficacy versus adalimumab, MONARCH additionally demonstrated improvement in functional outcomes for patients (table 2). Taken together, the robust MONARCH results further demonstrate that targeting IL-6R may be a preferred treatment option for patients who use biologics as monotherapy.
MONARCH was not without limitations. Although sarilumab plus MTX demonstrated superior radiographic results versus placebo plus MTX,24 the present study did not evaluate radiographic outcomes after sarilumab monotherapy compared with adalimumab monotherapy. Another limitation of this study is that it did not compare the efficacy of sarilumab monotherapy with sarilumab in combination with MTX. However, as patients intolerant to MTX were the primary intended target population, it would not be feasible to evaluate the addition of sarilumab to MTX in the context of this study.
Collectively, these data demonstrate that sarilumab improves signs and symptoms and functional disability of RA and is an appropriate, effective and superior monotherapy compared with TNF-α inhibition for patients who are unsuitable candidates for continued treatment with MTX due to intolerance or inadequate response.
Acknowledgments
Editorial assistance was provided under the direction of the authors by Gretchen Chidester, PhD, and Jennifer Rossi, MA, ELS, MedThink SciCom; Brandy L Bennett, PhD, and Michelle C DeSimone, PhD, DABT, Regeneron Pharmaceuticals and funded by Sanofi Genzyme and Regeneron Pharmaceuticals. The authors thank the patients who participated in this study, the co-investigators for their contribution to the study, staff at the participating centres and the following contributors for providing support: Sophie Bath-Ducrot and Sandra Hankins, for assistance in coordination and execution of the study; Stefano Fiore, for assistance in study design; Petur Wung, for assistance in data acquisition; Alexander Boddy, for assistance in study design, data acquisition and analysis and interpretation of the data and Chieh-I Chen and Clare Proudfoot for assistance in data analysis and interpretation.
References
Footnotes
Handling editor Tore K Kvien
Contributors GRB, YL, RP and JvA were involved in the conception and design of the study. GRB, YL, RP, DB, JIV and EBL were involved in data acquisition. GRB, YL, RP, JvA, EKM, NMHG, HvH, DB, JIV and EBL were involved in data analysis and interpretation. GRB, YL and JvA were involved in manuscript drafting. GRB, YL, RP, JvA, EKM, NMHG, HvH, DB, JIV and EBL were involved in critically revising the manuscript and approved the final version.
Funding This study was sponsored by Sanofi Genzyme and Regeneron Pharmaceuticals.
Competing interests GRB has received research grants or consulting fees from AbbVie, Bristol-Myers Squibb, MedImmune, Merck, Pfizer, Roche and UCB and has participated in speakers' bureaus for AbbVie, Bristol-Myers Squibb, Merck, Pfizer, Roche and UCB. EBL has acted as a consultant to Pfizer. JIV has received speaker fees from Roche, Bristol-Myers Squibb and Pfizer and has participated in speakers' bureaus for Bristol-Myers Squibb. YL, RP, HvH and DB are employees of Sanofi Genzyme and may hold stock and/or stock options in the company. JvA, EKM and NMHG are employees of Regeneron Pharmaceuticals and may hold stock and/or stock options in the company.
Ethics approval Individual Study Sites’ Institutional Review Boards.
Provenance and peer review Not commissioned; externally peer reviewed.