Article Text

Abstract
Objective To demonstrate pharmacokinetic equivalence of CT-P10 and innovator rituximab (RTX) in patients with rheumatoid arthritis (RA) with inadequate responses or intolerances to antitumour necrosis factor agents.
Methods In this randomised phase I trial, patients with active RA were randomly assigned (2:1) to receive 1000 mg CT-P10 or RTX at weeks 0 and 2 (alongside continued methotrexate therapy). Primary endpoints were area under the serum concentration–time curve from time zero to last quantifiable concentration (AUC0–last) and maximum serum concentration after second infusion (Cmax). Additional pharmacokinetic parameters, efficacy, pharmacodynamics, immunogenicity and safety were also assessed. Data are reported up to week 24.
Results 103 patients were assigned to CT-P10 and 51 to RTX. The 90% CIs for the ratio of geometric means (CT-P10/RTX) for both primary endpoints were within the bioequivalence range of 80%–125% (AUC0–last: 97.7% (90% CI 89.2% to 107.0%); Cmax: 97.6% (90% CI 92.0% to 103.5%)). Pharmacodynamics and efficacy were comparable between groups. Antidrug antibodies were detected in 17.6% of patients in each group at week 24. CT-P10 and RTX displayed similar safety profiles.
Conclusions CT-P10 and RTX demonstrated equivalent pharmacokinetics and comparable efficacy, pharmacodynamics, immunogenicity and safety.
Trial registration number NCT01534884.
- Rheumatoid Arthritis
- Pharmacokinetics
- DMARDs (biologic)
- B cells
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Rituximab is an anti-CD20 monoclonal antibody that exerts its effects via depletion of CD20+ B-cells.1 Following clinical trials,2 ,3 innovator rituximab (RTX) was approved for use in combination with methotrexate (MTX) in patients with rheumatoid arthritis (RA) with an inadequate response or intolerance to antitumour necrosis factor (TNF) agents.
CT-P10 is a candidate biosimilar of RTX. CT-P10 and RTX share an identical primary structure, as well as highly similar higher-order structures, post-translational modifications and in vitro biological activities (see online supplementary material A for comparative biological data). A key step in biosimilar development is to demonstrate pharmacokinetic (PK) equivalence to the innovator biologic (or ‘reference product’).4 ,5 We report the results of a phase I trial that assessed the PK equivalence—and additionally compared the efficacy, pharmacodynamics (PD), immunogenicity and safety—of CT-P10 and RTX.
supplementary materials
Patients and methods
Patients
Patients were aged 18–75 years, had active RA despite MTX treatment, and had inadequately responded or been intolerant to previous treatment with anti-TNF agents (see online supplementary material B and C for details of study methods).
Study design and treatment
This multinational, randomised, parallel-group, double-blind phase I trial was performed between March 2012 and May 2013 in 55 centres in eight countries in Europe, Asia and Latin America (ClinicalTrials.gov identifier NCT01534884).
On day 0, patients were randomly assigned 2:1 to receive intravenous infusions of 1000 mg CT-P10 (CELLTRION, Incheon, Korea) or 1000 mg RTX (Roche, Welwyn Garden City, UK) at day 0 (week 0) and week 2.
The main objective was to demonstrate PK equivalence between CT-P10 and RTX as assessed using the primary endpoints, area under the serum concentration–time curve from time zero to last quantifiable concentration (AUC0–last) and maximum serum concentration after second infusion (Cmax).
Statistical analyses
The primary statistical analysis was a comparison of AUC0–last and Cmax between CT-P10 and RTX groups, stratifying for region and prior anti-TNF agent status at baseline. The PK of the two drugs were to be considered equivalent if 90% CIs for the ratio of geometric means (CT-P10/RTX) of both primary endpoints fell within the bioequivalence range (80%–125%).
Results
Patients
Overall, 154 patients were enrolled and randomised to CT-P10 (N=103) or RTX (N=51) (see online supplementary material D). Demographics and baseline scores for disease activity assessments were similar between groups (table 1). Systemic corticosteroid use was also similar in the CT-P10 and RTX groups (mean daily dose (prednisolone equivalent): 6.30 and 6.69 mg, respectively, at baseline; 6.24 and 6.72 mg at week 24).
Demographics and baseline disease characteristics, including baseline scores for disease activity assessments (safety population)*
Pharmacokinetics
The PK of CT-P10 and RTX were equivalent since 90% CIs for the ratio of geometric means (CT-P10/RTX) for both AUC0–last and Cmax were within the bioequivalence range (table 2; see online supplementary material E). All secondary PK endpoints were also highly similar between groups (table 2). Geometric means of AUC0–last and Cmax were higher in patients negative for antidrug antibodies (ADA) than in those with at least one post-treatment ADA-positive result. Both endpoints, however, were equivalent across treatment groups in each ADA subset (see online supplementary material F).
PK endpoints (PK population)
To rule out effects of intrapatient variability on PK, individual patient log values of the two primary endpoints were plotted against each other. Positive high correlation between log(AUC0–last) and log(Cmax) was observed in both treatment groups. Interpretation of the same analysis by ADA subset was limited by the small number of patients; however, similar correlation trends were also observed (see online supplementary material G).
Efficacy
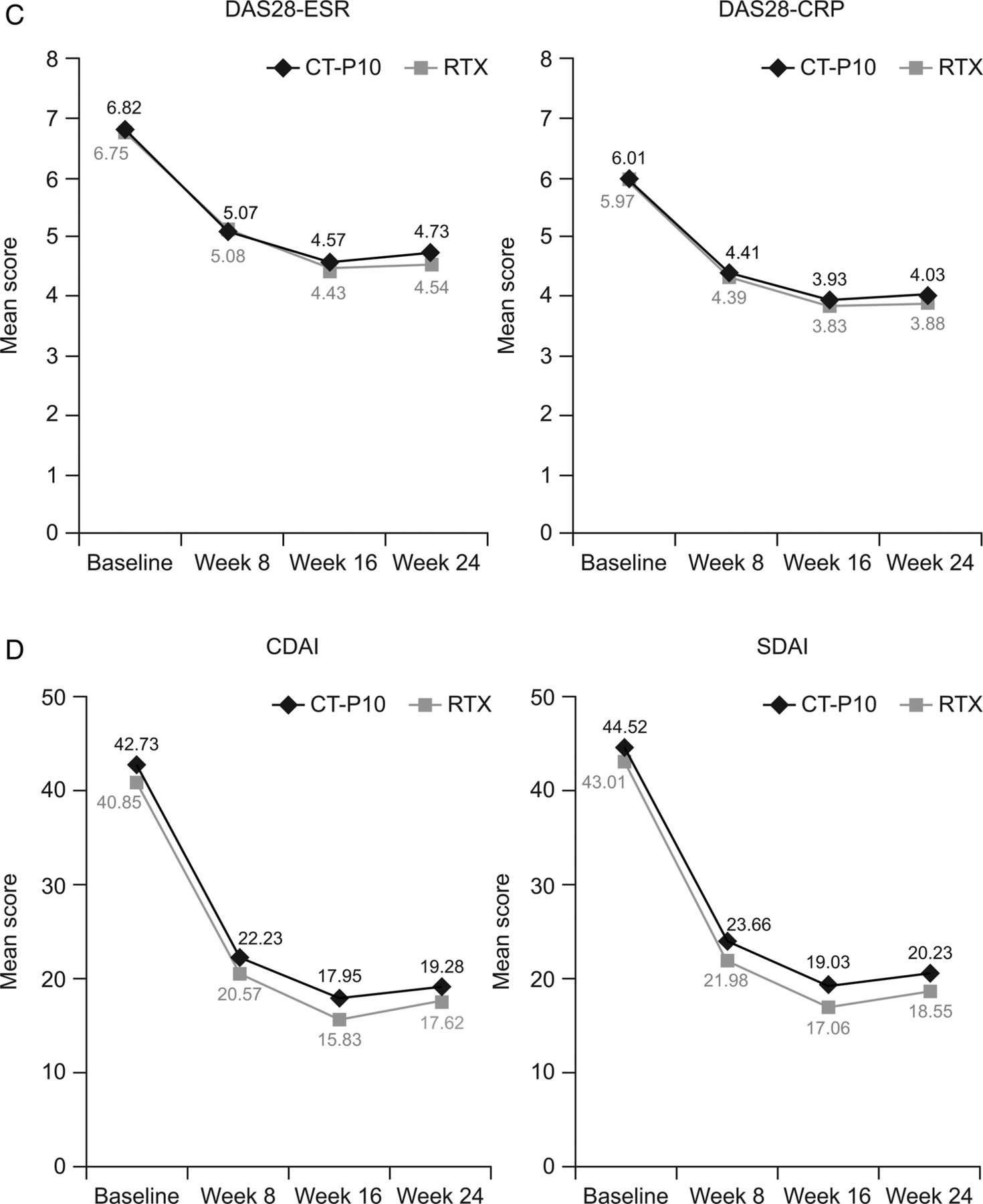
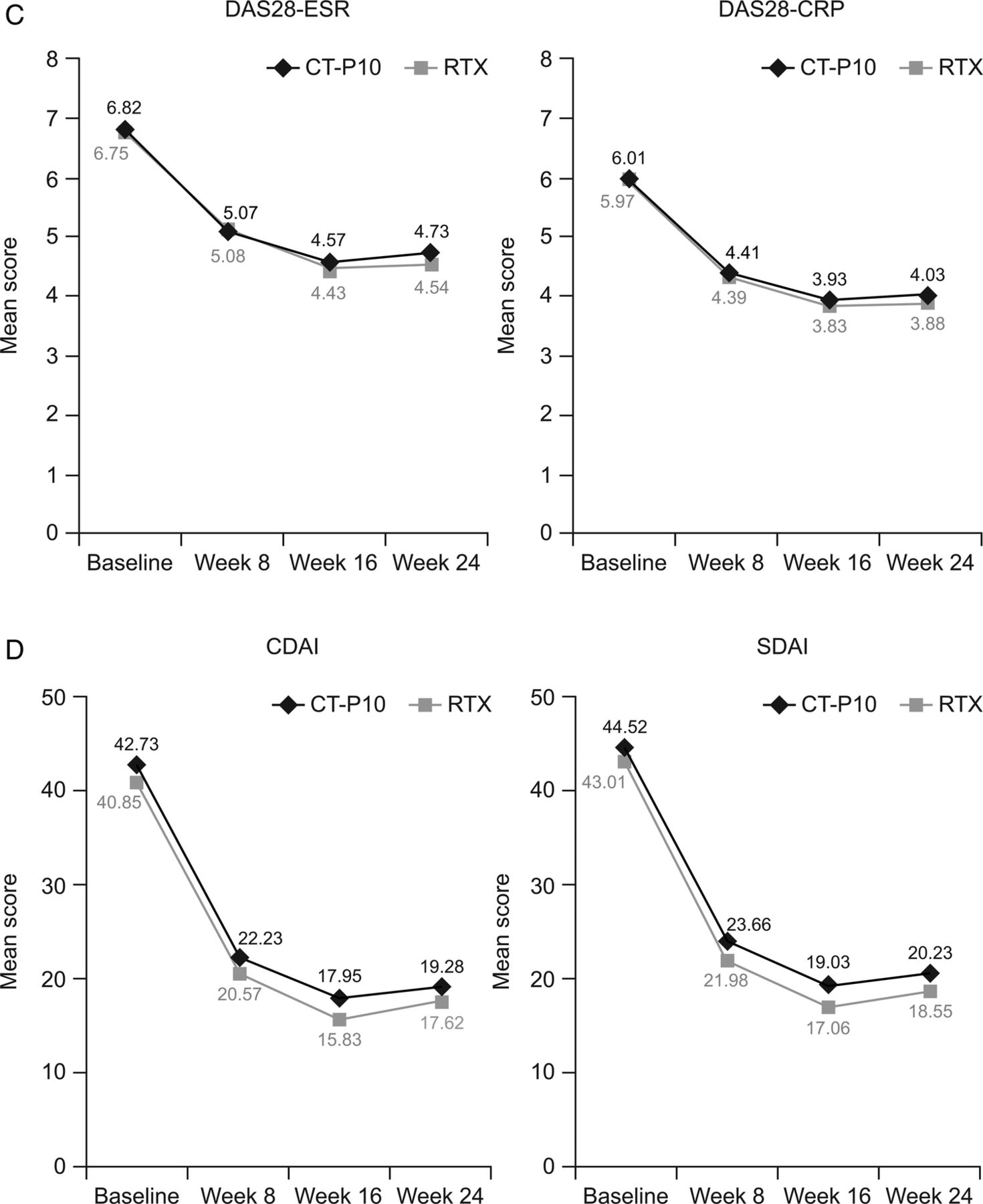
Efficacy was similar between treatments groups, with improvements from baseline observed in all endpoints. American College of Rheumatology (ACR) response rates were highly similar between groups (figure 1A). Mean changes from baseline at week 24 in all components of the ACR response were also highly similar between CT-P10 and RTX (see online supplementary material H), as was median (25th percentile, 75th percentile) time-to-onset of ACR20 response (58.0 (57.0 to 116.0) days and 60.0 (57.0 to 169.0) days for CT-P10 and RTX, respectively). The proportion of patients achieving good or moderate European League Against Rheumatism (EULAR) responses, and decreases in mean scores from baseline in Disease Activity Score in 28 joints (DAS28), Clinical Disease Activity Index and Simplified Disease Activity Index, were comparable between the two groups (figure 1B–D). At week 24, mean changes from baseline in DAS28 were not significantly different between the CT-P10 and RTX groups (DAS28-C reactive protein (CRP): −1.946 and −2.047, respectively (p=0.66; 95% CI for the difference in change from baseline: −0.36 to 0.43); DAS28-erythrocyte sedimentation rate, −2.065 and −2.147 (p=0.73; 95% CI −0.39 to 0.56)). Improvements in the 36-item Short Form Health Survey (SF-36) and physical component and mental component summary scores were of similar magnitude in the two treatment groups.

(A) Proportion of patients with an ACR20, ACR50 and ACR70 response. (B) Proportion of patients achieving a good or moderate EULAR response. (C) Mean DAS28 over time. (D) Mean CDAI and SDAI over time. Data are shown for the efficacy population (CT-P10, N=100; RTX, N=48). ACR, American College of Rheumatology; ACR20, 20% response according to the ACR criteria for improvement; ACR50, 50% response according to the ACR criteria for improvement; ACR70, 70% response according to the ACR criteria for improvement; CDAI, Clinical Disease Activity Index; CRP, C reactive protein; DAS28, Disease Activity Score in 28 joints; ESR, erythrocyte sedimentation rate; EULAR, European League Against Rheumatism; RTX, rituximab; SDAI, Simplified Disease Activity Index.
{kind=link}
{kind=link}
Continued
At week 24, ACR20 response rates in the CT-P10 and RTX groups, respectively, were 61.1% and 62.5% in patients with ADAs and 67.5% and 75.0% in those without ADAs. There was no statistically significant difference in ACR20 response rate between ADA subsets in each treatment group. Similarly, there were no significant differences in good or moderate EULAR response rates between ADA subsets in either treatment group.
PD and immunogenicity
Rapid and complete depletion of CD19+ peripheral B-cells was observed after infusion of CT-P10 or RTX (see online supplementary material I). No significant reductions in immunoglobulin levels were observed (see online supplementary material J).
ADAs were detected in 18 (17.6%) and 9 (17.6%) patients in the CT-P10 and RTX groups, respectively, at week 24; neutralising antibodies were detected at week 24 in 2 (2.0%) and 1 (2.0%) patient, respectively.
Safety
Adverse events occurred in 52 (51.0%) and 38 (74.5%) patients in the CT-P10 and RTX groups, respectively, and serious adverse events in 5 (4.9%) and 3 (5.9%). Infusion-related reactions occurred in 17 (16.7%) patients in the CT-P10 group and 10 (19.6%) in the RTX group. These reactions occurred after the first and second infusions in 15 (14.7%) and 4 (4.0%) patients, respectively, in the CT-P10 group and in 10 (19.6%) and no patients in the RTX group. Most infusion-related reactions were grade 1 or 2 in severity; one was grade 3 (headache in the CT-P10 group; resolved without treatment). Infections occurred in 24 (23.5%) and 13 (25.5%) patients in the CT-P10 and RTX groups, respectively. There were no life-threatening (grade 4) adverse events or deaths.
In each treatment group, there were no statistically significant differences in the incidence of serious adverse events, infusion-related reactions or infections between patients with and without ADAs at week 24 (p>0.05 for all comparisons). Online supplementary material K reports additional safety data.
Discussion
The primary aim of this study was to demonstrate PK bioequivalence between CT-P10 and RTX in patients with RA. Unlike most phase I trials, the study also assessed efficacy. Another unusual feature was the 2:1 randomisation scheme. Original sample size calculations for the primary endpoint showed 50 patients were needed in each group considering a 20% drop-out rate. However, to allow further assessment of CT-P10 safety, we recruited another 50 patients to the CT-P10 group; this also led to an increase in study power.
The primary endpoints (AUC0–last and Cmax) were equivalent between CT-P10 and RTX as the 90% CIs for the ratios of their geometric mean values were within the predefined equivalence margins. These margins (80%–125%) were considered appropriate, and tighter margins not feasible, due to the broad therapeutic window of RTX and high interpatient variability in AUC previously observed with RTX.6–8 In both groups, mean AUC0–last and Cmax were similar to previous reports for RTX in patients with RA (AUC0–last (μg hour/mL): 200 238 and 203 783 for CT-P10 and RTX, respectively, vs 190 320–242 000;6 ,7 Cmax (μg/mL): 466 and 478, respectively, vs 355–453).7 ,8 Secondary PK outcomes were also highly similar between CT-P10 and RTX.
Highly similar efficacy data were observed between the two treatments groups. ACR/EULAR responses and DAS28 scores in both groups were similar to those observed with RTX in the REFLEX and DANCER trials.2 ,3 Decreases in CRP levels (mg/dL) at week 24 were slightly lower in this study than in REFLEX (−2.1 for RTX in REFLEX vs −0.8 and −1.1 for CT-P10 and RTX, respectively, here). This is likely a reflection of the higher baseline CRP level in REFLEX (3.7 vs 1.8 and 2.1, respectively).2 In the current study, no significant effects of ADAs on efficacy were observed although numerical reductions in response rates (of around the same magnitude) were noted in both treatment groups.
B-cell kinetic observations were as expected while the proportion of ADA-positive patients (17.6% in each group at week 24) was higher than observed in previous RTX studies (12.7%).8 This may have been because the electrochemiluminescent immunoassay used is more sensitive than the ELISA employed in most RTX trials.9 False ADA-positive results were observed in some patients at baseline, probably due to assay interference by CD20+ B-cell membrane fragments.10
Safety findings for CT-P10 and RTX were comparable. The incidence of infusion-related reactions declined with subsequent infusions in both groups. That no such reactions were reported after the second infusion in the RTX group, compared with a rate of around 9% in other RTX trials,11 was likely a random finding. Presence of ADAs did not affect the safety of either drug.
In this study, CT-P10 and RTX demonstrated equivalent PK and comparable efficacy, PD, safety and immunogenicity up to week 24 in patients with RA. The results provide a clear rationale for future studies of CT-P10.
supplementary data
Acknowledgments
The authors wish to thank the patients and study personnel who made this trial possible and the study investigators—Germany: Spieler W, Rech HJ, Braun J; Poland: Brzezicki J, Ruzga Z, Piotrowski M, Tlustochowicz W, Porawska W; Mexico: Simon Campos JA, Ramos-Remus C, Salazar-Paramo M; Russia: Raskina T, Vinogradova I; Brazil: Scheinberg M, Scotton A; Korea: Park KS, Jun JB, Park YB, Lee SS; Spain: Rey JS, Fernandez-Nebro A, Navarro Blasco FJ; Ukraine: Polyvoda S, Polyakov A. Medical writing support on this manuscript was provided by Louise Niven DPhil at Aspire Scientific Limited (Bollington, UK) and was funded by CELLTRION (Incheon, Korea).
Footnotes
Handling editor Tore K Kvien
Contributors DHY, SJL, SYL and TSK were involved in conception and design of the study, acquisition of data and/or analysis and interpretation of data; drafting of the manuscript and revising it critically for important intellectual content and final approval of the version to be published. WP, C-HS and SCS were involved in the conception and design of the study; drafting of the manuscript and revising it critically for important intellectual content and final approval of the version to be published. SJ, FFCM, PH, PW, EYL, FGM-R, PS, SR, MS, VK, DHS, LM, MJL and J-YC were involved in the acquisition of data; drafting of the manuscript and revising it critically for important intellectual content and final approval of the version to be published. The project management, clinical and medical monitoring, pharmacovigilance, data management, analysis of pharmacokinetic data, biostatistical analysis and medical writing were performed under contract with PPD in collaboration with CELLTRION.
Funding The study was sponsored by CELLTRION (Incheon, Korea).
Competing interests DHS, EYL, FFCM, FGM-R, J-YC, LM, MJL, MS, PS, SJ, SR and VK have received grants from CELLTRION during the conduct of the study. PH has received grants during the conduct of the study and honoraria for talks outside the submitted work from CELLTRION. C-HS, DHY and WP have received personal fees during the conduct of the study and grants outside the submitted work from CELLTRION. SCS has received personal fees during the conduct of the study from CELLTRION. PW has received grants from CELLTRION during the conduct of the study and personal fees from Roche, Abbvie, Angelini, Egis, Merck Sharp & Dohme and Pfizer, outside the submitted work. SJL, SYL and TSK are employees of CELLTRION.
Patient consent Obtained.
Ethics approval The protocol was reviewed and approved by each site's institutional review board or independent ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data available for this paper are included in the manuscript and online supplementary appendices.