Article Text

Abstract
Objective To demonstrate pharmacokinetic (PK) similarity of biosimilar candidate ABP 501 relative to adalimumab reference product from the USA and European Union (EU) and evaluate safety, tolerability and immunogenicity of ABP 501.
Methods Randomised, single-blind, single-dose, three-arm, parallel-group study; healthy subjects were randomised to receive ABP 501 (n=67), adalimumab (USA) (n=69) or adalimumab (EU) (n=67) 40 mg subcutaneously. Primary end points were area under the serum concentration-time curve from time 0 extrapolated to infinity (AUCinf) and the maximum observed concentration (Cmax). Secondary end points included safety and immunogenicity.
Results AUCinf and Cmax were similar across the three groups. Geometrical mean ratio (GMR) of AUCinf was 1.11 between ABP 501 and adalimumab (USA), and 1.04 between ABP 501 and adalimumab (EU). GMR of Cmax was 1.04 between ABP 501 and adalimumab (USA) and 0.96 between ABP 501 and adalimumab (EU). The 90% CIs for the GMRs of AUCinf and Cmax were within the prespecified standard PK equivalence criteria of 0.80 to 1.25. Treatment-related adverse events were mild to moderate and were reported for 35.8%, 24.6% and 41.8% of subjects in the ABP 501, adalimumab (USA) and adalimumab (EU) groups; incidence of antidrug antibodies (ADAbs) was similar among the study groups.
Conclusions Results of this study demonstrated PK similarity of ABP 501 with adalimumab (USA) and adalimumab (EU) after a single 40-mg subcutaneous injection. No new safety signals with ABP 501 were identified. The safety and tolerability of ABP 501 was similar to the reference products, and similar ADAb rates were observed across the three groups.
Trial registration number EudraCT number 2012-000785-37; Results.
- Pharmacokinetics
- TNF-alpha
- Anti-TNF
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
ABP 501 is being developed as a biosimilar to adalimumab (Humira); biosimilars are similar versions of approved branded biologics. Adalimumab is a recombinant IgG1 monoclonal antibody (mAb) that binds to the tumour necrosis factor α (TNFα) cytokine to block its interaction with the p55 and p75 cell surface TNF receptors. Adalimumab has been shown to reduce disease symptoms for all approved therapeutic indications, to inhibit the progression of structural damage in rheumatoid arthritis, juvenile idiopathic arthritis and psoriatic arthritis, and to induce and maintain clinical remission in adults with moderately to severely active Crohn's disease, ulcerative colitis, plaque psoriasis and ankylosing spondylitis.1 ,2
Although targeted biologics, including adalimumab, have demonstrated safety and efficacy in the treatment of autoimmune disorders, their widespread application may be limited due to their high cost.3 With the expiration of patents on several biologics and more expirations anticipated soon, there is growing interest in the development of biosimilars, which are expected to be less expensive therapeutic alternatives to branded biologics.4 The US Food and Drug Administration has developed an abbreviated and expedited pathway—the 351(k) pathway—for the approval of biosimilars5 and has released guidance for industry to support their development.6–8 The European Union (EU) previously published guidelines for the development and approval of biosimilars.9
According to the guidance, biosimilars should be ‘highly similar’ to the reference product with respect to quality attributes, notwithstanding minor differences in clinically inactive components, and should have no clinically meaningful differences with respect to safety, purity and potency. Biosimilar guidance recommends a totality-of-evidence approach focused on stepwise development of a proposed biosimilar, which starts with the demonstration of similarity to the reference product based on analytical characterisation, pharmacological activity, pharmacokinetics (PKs) and pharmacodynamics (PD).6 ,9 This is followed by comparative clinical evaluation of the proposed biosimilar candidate in a sensitive population to demonstrate similar efficacy and safety at the same approved dose and route of administration as the reference product. Demonstration that the proposed biosimilar is not more immunogenic than the reference product is also required.6 ,10
There are several biosimilar candidates for inflammatory disease currently under development, including: SB2 infliximab biosimilar, SB4 etanercept biosimilar, SB5 adalimumab biosimilar (Samsung), BI 695501 adalimumab biosimilar (Boehringer Ingelheim), CT-P10 rituximab biosimilar (Celltrion), ABP 501 adalimumab biosimilar, ABP 798 rituximab biosimilar and ABP 710 infliximab biosimilar (Amgen). An infliximab biosimilar, CT-P13 (Remsima/Inflectra, Celltrion), has been approved for use in the EU, USA and Canada.
ABP 501 is a fully human recombinant monoclonal antibody with the same amino acid sequence, pharmaceutical form and dosage strength as adalimumab. It is, however, not formulated with the same excipients as adalimumab and includes different buffer components and stabilisers. ABP 501 is being developed for the same indications, dosages and route of administration as approved for adalimumab; the drug product is supplied as a sterile, preservative-free solution for administration by subcutaneous (SC) injection. Biologic products (proteins and their formulations) may have different bioavailability; the purpose here is to demonstrate PK similarity between the biosimilar and its reference product.
The totality of evidence available to date suggests that ABP 501 is similar to adalimumab. Results from analytical studies that evaluated identity, general properties, primary and higher-order structure, carbohydrate structure, isoelectric profile, purity and impurities, and thermal-forced degradation profiles have confirmed ABP 501 to be structurally similar to adalimumab.11 Results from functional characterisation studies have demonstrated that ABP 501 and adalimumab have similar binding affinity to TNFα and comparable inhibition of TNFα activities in vitro.12 Additionally, ABP 501 and adalimumab have shown comparable induction of effector functions as seen from the results of antibody-dependent cell-mediated cytotoxicity and complement-mediated cytotoxicity assays.12 ABP 501 has also been shown to be similar to adalimumab with respect to binding to a panel of Fc receptors, including FcγRIa, FcγRIIa, FcγRIIIa (158V), FcγRIIIa (158F) and FcRn.12 The similarity in binding to FcRn is particularly relevant because binding of mAbs to FcRn affects their clearance.
This phase I study was conducted to evaluate the PK similarity of ABP 501 with adalimumab. The adalimumab comparator product was sourced from the USA and the EU; both are manufactured by the same company and are structurally and functionally equivalent.11 The purpose of this phase I study was to demonstrate the PK equivalence of ABP 501 to adalimumab (USA) and adalimumab (EU) in healthy subjects after a single 40-mg SC dose. The secondary objectives were to assess safety, tolerability and immunogenicity.
Methods
Subjects
Healthy adults, 18–45 years of age with a body mass index of 18–30 kg/m2 at screening were eligible. Subjects were excluded if they had an infection (bacterial, viral, parasitic, systemic fungal or other opportunistic pathogen) ≤30 days of investigational product (IP) administration, had tuberculosis (latent or active) ≤6 months of screening, or reported a current malignancy or a malignancy ≤5 years (with the exception of excised nonmelanoma skin cancer), were receiving or had received any investigational drug or device ≤30 days (USA) or 90 days (EU) or five half-lives (whichever was longer), or had previously received adalimumab or any product considered to be a biosimilar to adalimumab. All subjects provided written informed consent.
Study design
This was a randomised, single-blind, single-dose, three-arm, parallel-group study conducted at one clinical pharmacology unit (CPU) in the USA and one CPU in the EU using adalimumab sourced from each region. The study was conducted according to the Declaration of Helsinki and in compliance with good clinical practices and EU clinical trial directives. The study protocol was approved by an independent ethics committee or institutional review board at each site prior to study initiation. A total of 203 subjects were enrolled in the study. Screening occurred ≤28 days before dosing. Eligible subjects were admitted to the CPU on day 1 and randomised in a ratio of 1:2 by the two regions such that the ratio of subjects to receive ABP 501 40 mg (Amgen), adalimumab 40 mg (USA; AbbVie) or adalimumab 40 mg (EU; AbbVie) was the same before dosing on day 1. Subjects were discharged from the CPU on day 2, and returned to the CPU over the next 63 days for safety evaluations and blood sampling for PK and antidrug antibody (ADAb) assessments. Subjects were monitored throughout the study for adverse events (AEs), clinical laboratory results, concomitant medication use and vital signs. Samples for PK assessments were collected on days 1 (predose, 1, 4, 8, 12 hours post dose), 2, 3, 4, 5, 6, 7, 8, 9, 11, 14, 16, 22, 29, 36, 43, 50, 57 and 63 (end of study). ADAb tests were performed at prespecified visits, including days 1 (predose), 16, 29 and 63. Serum concentrations of adalimumab and ABP 501 were determined using validated electrochemiluminescent (ECL) assays. The ECL assay method was based on the Meso Scale Discovery platform using an anti-ID mAb to capture ABP 501 and adalimumab reference product from test samples and a second ruthenium-labelled anti-ID to detect the bound test products. The binding ADAbs were determined using a validated ECL assay that detects all antibody classes.13–15 A cell-based assay was used for detecting neutralising ADAbs using a TNFα-responding cell line that results in a TNFα-induced phosphorylation of nuclear factor κB (NFκB). TNFα was added to induce phosphorylation of NFκB in the cell line, which was inhibited by addition of known amount of drug. In the presence of the neutralising ADAbs the phosphorylation of NFκB is restored. Briefly, cells were incubated with the drug, serum sample and recombinant human TNFα and lysed, followed by measurement of the phospho-NFκB. This bridging assay could detect all subclasses of bivalent or multivalent ADAbs; the ADAb response to ABP 501 and adalimumab occurred with a wide range of subclasses. Whereas the assay may not detect the monovalent IgG4 isotype, there are other subclasses of ADAbs present in much higher concentrations in the serum than monovalent IgG4 antibodies. The assay was validated with a tolerance of 25 µg/mL of drug; the highest observed maximum observed concentration (Cmax) in this study was below 6.0 µg/mL. Therefore, drug interference was not expected from the collected samples. Additionally, the neutralising antibody cell-based bioassay developed for this study was expected to detect all classes of antibodies that inhibit the biological activity of the drug, including monovalent IgG4 subclass antibodies. Assay validation included determination of drug tolerance by adding excess drug as described by Shankar et al.16 The specificity of the assay was tested with a rabbit-positive control; other anti-TNF biologics do not interfere with the assay. The assays were validated using both adalimumab (USA) and adalimumab (EU) and shown to be of equal sensitivity to both materials. Serum concentrations of each of the three test molecules were determined and summarised for all subjects who received any IP and had at least one reported serum concentration of adalimumab reference products or ABP 501.
Investigational product
All subjects receiving adalimumab (USA) or adalimumab (EU) received the IP from a single lot for each region. All subjects in the ABP 501 study arm received IP from a single lot in each region although different lots were used between the USA and the EU.
Study objectives
The primary objective of this study was to demonstrate that the PK of ABP 501 is similar to adalimumab (USA) and adalimumab (EU), as assessed by the area under the serum concentration-time curve (AUC) from time 0 extrapolated to infinity (AUCinf) and the Cmax. Secondary objectives included assessment of the time to Cmax (tmax), AUC from time 0 to the last quantifiable concentration (AUClast) of ABP 501 compared with adalimumab (USA) and adalimumab (EU), as well as evaluation of safety, tolerability and immunogenicity.
Statistical analyses
Sample size was estimated using previous bioavailability studies with adalimumab; ∼198 subjects were to be enrolled in this three-arm study (66 per arm). The PK parameter population consisted of all subjects with an evaluable adalimumab or ABP 501 serum concentration-time profile; this population was used for the primary analysis of PK equivalence. Sensitivity analyses were planned to assess PK equivalence using the per-protocol PK parameter population, and in subpopulations of region and binding ADAb-negative and ADAb-positive subjects. The safety analysis set consisted of all subjects who received any amount of IP.
PK parameters were calculated using non-compartmental techniques (WinNonlin Professional Network Edition, V.5.2, Pharsight, St. Louis, Missouri, USA) for all subjects with an evaluable adalimumab or ABP 501 serum concentration-time profile. PK similarity was assessed by comparing the 90% CIs for the geometrical mean (GM) test-to-reference ratios for AUCinf, Cmax, and AUClast with the protocol-specified bioequivalence criteria of 0.80 and 1.25; AUClast was evaluated to fully assess exposure to the IP. Prior to statistical modelling, PK parameters were log-transformed. Point estimates and 90% CIs for the mean difference in logarithmic PK parameters were estimated using an analysis of covariance model for comparisons of ABP 501 and adalimumab (USA), ABP 501 and adalimumab (EU), and adalimumab (USA) and adalimumab (EU), adjusted for region (USA and EU) and weight. Point estimates and 90% CIs for GM ratios (GMRs) were then calculated by transforming back to the original scale. To establish PK similarity, the 90% CIs of the GMRs were to be within the standard equivalence criteria of 0.80 and 1.25.17 ,18 Serum concentrations and PK parameters were also summarised using descriptive statistics by treatment group.
Safety and immunogenicity of ABP 501 compared with adalimumab were evaluated through descriptive summaries of AEs, clinical laboratory tests, physical examination, vital signs, ECG, concomitant medications and incidence of ADAb.
Results
Subject disposition and characteristics
Subject disposition is summarised in figure 1. All 203 randomised subjects were dosed and were included in the safety and PK parameter populations for analyses; all but seven subjects completed the study as planned. Reasons for early discontinuation included withdrawal of consent, non-compliance with study protocol, loss to follow-up and one serious AE (SAE) of dermoid cyst that was considered to be unrelated to study treatment. A total of 67 subjects received ABP 501 in the USA and EU, 69 subjects received adalimumab (USA) and 67 subjects received adalimumab (EU). Data from six of the seven subjects who did not complete the study were included in the PK analysis and data from all seven subjects were used in the ADAb analysis.

Subject disposition.
A summary of baseline characteristics is provided in table 1. Baseline characteristics were comparable between treatment groups.
Summary of demographic data and baseline characteristics (safety population)
Pharmacokinetics
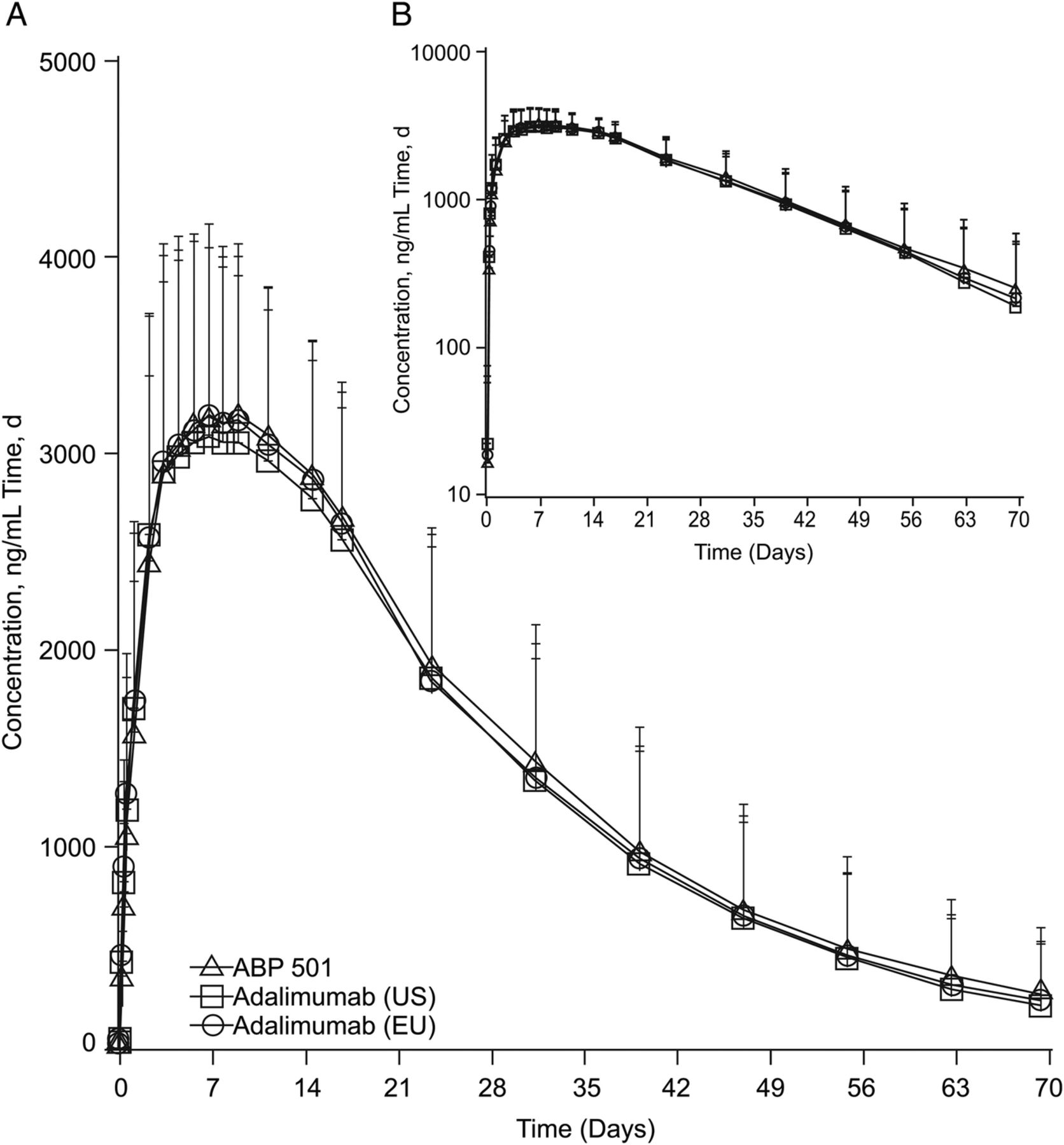
The mean serum concentration-time profiles after a single SC injection of ABP 501, adalimumab (USA) and adalimumab (EU) were similar among the three groups (figure 2). Peak concentrations were observed approximately 1 week after injection, after which concentrations declined in a monophasic manner.
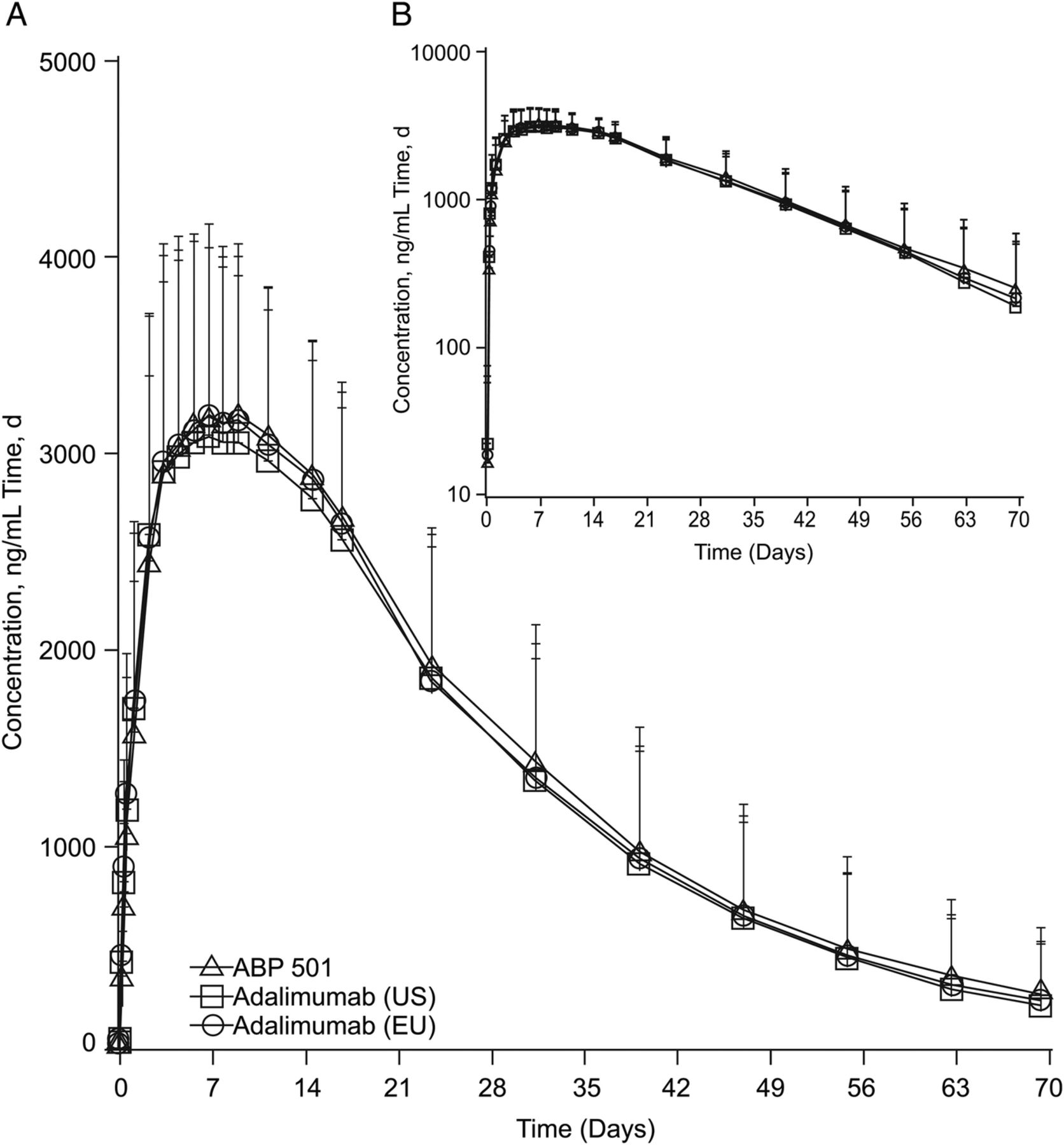
Mean serum ABP 501, adalimumab (USA) and adalimumab (EU) concentration-time profiles (A) Linear scale. (B) Semilogarithmic scale. Error bars=SD.
The GMs of PK parameters after a single SC injection of ABP 501, adalimumab (USA) and adalimumab (EU) were similar among the three groups. Likewise, both peak and overall exposures were similar across the three treatment groups, as was the tmax.
The 90% CIs for the GMRs of Cmax, AUCinf and AUClast were fully contained within 0.80 to 1.25, confirming the PK equivalence between ABP 501 and adalimumab (USA) and adalimumab (EU), and between adalimumab (USA) and (EU) (table 2).
Ratio of adjusted least squares geometrical means of ABP 501, adalimumab (USA) and adalimumab (EU) (PK parameter population)
Safety
There were no deaths and no treatment-emergent SAEs or treatment-emergent AEs (TEAEs) leading to discontinuation from the study that were considered by the investigators to be related to the study drug. One subject in the adalimumab (EU) treatment group reported an SAE of dermoid cyst which was considered unrelated to the study drug; the subject was withdrawn from the study due to this SAE. TEAEs of any grade were reported for 58.2%, 47.8% and 68.7% of subjects in the ABP 501, adalimumab (USA) and adalimumab (EU) groups, respectively. The most frequently reported TEAEs by preferred term were headache, oropharyngeal pain, sinus congestion, nasopharyngitis and nausea (table 3). TEAEs that were assessed as possibly or probably related to the study drug were either mild or moderate in intensity and were reported for 35.8%, 24.6% and 41.8% of subjects in the ABP 501, adalimumab (USA) and adalimumab (EU) groups, respectively. No clinically relevant changes or trends in clinical laboratory tests, ECG, vital signs and physical examinations were observed during the study.
Summary of adverse events (safety population)
Immunogenicity
All samples were tested against ABP 501, adalimumab (USA) and adalimumab (EU). The ECL assay sensitivity for ADAbs was approximately 0.02 μg/mL in presence of 25 μg/mL drug. The drug concentrations were well below the assay drug tolerance in all subjects. Antibodies developed by day 16 in some subjects; most remained positive until the end of study (table 4).
Antidrug antibody incidence during the study
No pre-existing ADAbs were detected at baseline; all ADAbs detected during the study developed after dosing with ABP 501 or adalimumab. A total of 36 (54%), 38 (55%) and 45 (67%) subjects in the ABP 501, adalimumab (USA) and adalimumab (EU) treatment groups, respectively, developed binding ADAbs at some point during the study (table 4). Neutralising antibodies were detected in 12 (18%), 15 (22%) and 14 (21%) subjects in the ABP 501, adalimumab (USA) and adalimumab (EU) treatment groups, respectively.
As shown in table 5, PK parameters were similarly affected by binding ADAb status for the three treatments. Overall exposure was approximately 20–30% lower for all three treatments in ADAb-positive subjects compared with ADAb-negative subjects. The shorter terminal elimination half-life (t½) in ADAb-positive subjects was consistent with lower exposure (figure 3). The average t½ values were 6–7 days in the ADAb-positive subjects compared with 12–15 days in ADAb-negative subjects. The mean study drug half-lives for ADAb-negative subjects in the ABP 501, adalimumab (USA) and adalimumab (EU) treatment groups were 371, 281 and 366 hours, respectively, compared with 151, 169 and 176 hours in ADAb-positive subjects.
Summary of ABP 501, adalimumab (USA) and adalimumab (EU) pharmacokinetic parameters by antidrug antibody status

{kind=link}
{kind=link}
{kind=link}
Individual PK profiles depicting longer t1/2 in ADAb-negative subjects for all three test products: ABP 501, adalimumab (USA) and adalimumab (EU). ADAb, antidrug antibody; PK, pharmacokinetic.
Discussion
The effectiveness of anti-TNF agents, including adalimumab, to control disease progression in approved indications is well established through clinical trials.1 ,2 ,19–22 Biosimilars have the potential to improve access to biologics by providing less expensive treatment options with safety and efficacy profiles similar to those of the reference product.23 This randomised, single-blind, single-dose, three-arm, parallel-group study assessed the PK similarity and safety, tolerability and immunogenicity of proposed biosimilar ABP 501 (40 mg SC) and adalimumab (USA) and adalimumab (EU) (40 mg SC) in healthy men and women.
Human PK and/or PD studies are fundamental components in supporting biosimilarity. Based on regulatory guidelines, selection of appropriate study populations (patients vs healthy subjects) for these studies should be scientifically justified.6 This phase I study enrolled healthy subjects, comprising a homogeneous population: they were immune-competent and were not receiving concomitant medications for the indication of interest, thus allowing a sensitive comparison of PKs for ABP 501 and adalimumab. Healthy subjects and patients with rheumatoid arthritis have been shown to display similar PKs for adalimumab.1 The general standard for PK studies to establish bioequivalence is the 90% CI for the GMR being within the prespecified acceptance range of 0.8 and 1.25 for overall exposure (eg, AUC).17 ,18 The European Medicines Agency requires that the proposed biosimilar product has equivalent exposure as the reference product; demonstrating similar exposure is a critical step in the abbreviated development programme for biosimilars.10 Following the recommended stepwise approach,6 demonstrating similar PK properties after establishing similar analytical/functional activity allows the proposed biosimilar to be moved directly into phase III studies (skipping phase II studies), because similar exposure data support the assumption that the clinical dose for the proposed biosimilar is known.10
In this phase I study, the primary end points, AUCinf and Cmax, were shown to be similar for ABP 501 and the adalimumab reference products. Additionally, the reference product sourced from both regions, adalimumab (USA) and adalimumab (EU), were shown to be similar. The 90% CIs for GMRs of Cmax, AUCinf and AUClast were fully contained within the standard prespecified criteria of 0.80–1.25, confirming the PK similarity among ABP 501, adalimumab (USA) and adalimumab (EU). PK similarity assessment may also be conducted in patients with active disease as in the PLANETAS (Programme evaLuating the Autoimmune disease iNvEstigational drug cT-p13 in AS [Ankylosing Spondylitis] patients) study of biosimilar infliximab.24 As outlined above, we evaluated the PK of ABP 501 in healthy volunteers as this is the most sensitive population to detect differences because of lack of immunosuppressive medications or underlying disease. Additionally, PK comparison in the CPU setting allowed more stringent inclusion and exclusion criteria that reduced variability in PK end points.
In this study, single doses of ABP 501, adalimumab (USA) and adalimumab (EU) administered to healthy subjects showed comparable safety and tolerability between the treatment groups. The incidence of ADAbs was similar among the three groups. Although the proportion of subjects with binding ADAb was higher in subjects exposed to adalimumab (EU), the magnitude of response was similar. The ADAb rates observed for the adalimumab (EU) treatment arm are within previously reported ranges.25 The incidence of ADAbs observed in this study was higher than reported in adalimumab package inserts,1 ,2 likely because the binding ADAb assay used in this study was designed for enhanced sensitivity.15 The high proportion of subjects with ADAb positivity observed in the binding assay may be due to the highly sensitive Meso Scale Discovery assay. The neutralising antibody incidence by treatment was similar for ABP 501 (18%), adalimumab (USA) (22%) and adalimumab (EU) (21%, table 4). In each of the three groups, an observable decrease in the study drug half-life was identified in ADAb-positive subjects compared with ADAb-negative subjects. Serum antibodies to adalimumab are associated with lower serum adalimumab concentration, resulting in diminished treatment response and therefore reduced efficacy in patients with rheumatoid arthritis.26–30 The decrease in study drug half-life in ADAb-positive subjects who received ABP 501 was similar to that observed in subjects who received adalimumab.
Based on the results of this phase I PK similarity study and the totality of evidence from the preclinical, analytical and functional similarity studies, pivotal clinical studies were designed to assess clinical biosimilarity of ABP 501 and adalimumab. The goal of these studies is to determine whether there are clinically meaningful differences between ABP 501 and adalimumab in terms of safety, efficacy and immunogenicity under the conditions of use approved for adalimumab and in accordance with the regulations and guidance for biosimilars development.
Conclusions
In this phase I study, after a single 40-mg SC injection, the PK of ABP 501 was similar to that of adalimumab (USA) and adalimumab (EU). The safety and tolerability of ABP 501 and adalimumab (USA) and adalimumab (EU) were similar, as were the rates of ADAbs.
Acknowledgments
The authors thank Ralf Brueckner, MD, ICON, USA for study oversight. Medical writing support was provided by Monica Ramchandani, PhD, Amgen and Anthony Maine, ICON Development Solutions, USA. Editorial support, funded by Amgen, was provided by Complete Healthcare Communications.
References
Footnotes
Handling editor Tore K Kvien
†MM is deceased.
Contributors PK: contributed to conduct of study, data interpretation, reporting of results and manuscript development. VC: contributed to study design, conduct of study, data interpretation, reporting of results and manuscript development. NZ: contributed to study design and conduct of study, planning and execution of statistical analysis, data interpretation, reporting of results and manuscript development. MM: contributed to immunogenicity testing, data interpretation and manuscript development. AK: contributed to immunogenicity testing, data interpretation, and manuscript revisions. RM: study design, interpretation of data, reporting of results, manuscript development and overall responsibility for the development activities of this programme at Amgen.
Funding This study was funded by Amgen.
Competing interests PK, VC, NZ, MM, AK and RM are employees and stockholders of Amgen.
Patient consent Obtained.
Ethics approval Each site's institutional review board or independent ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.