Article Text

Abstract
Objectives To investigate the efficacy and safety of switching from infliximab reference product (RP) to its biosimilar or maintaining biosimilar treatment in patients with ankylosing spondylitis (AS).
Methods This open-label extension study recruited patients with AS who completed a 54-week, randomised controlled study comparing CT-P13 with RP (PLANETAS). CT-P13 (5 mg/kg) was administered intravenously every 8 weeks from week 62 to week 102. Efficacy end points included the proportion of patients achieving Assessment of SpondyloArthritis international Society (ASAS)20. Antidrug antibodies (ADAs) were measured using an electrochemiluminescent method. Data were analysed for patients treated with CT-P13 in the main PLANETAS study and the extension (maintenance group) and those who were switched to CT-P13 during the extension study (switch group).
Results Overall, 174 (82.9%) of 210 patients who completed the first 54 weeks of PLANETAS and agreed to participate in the extension were enrolled. Among these, 88 were maintained on CT-P13 and 86 were switched to CT-P13 from RP. In these maintenance and switch groups, respectively, ASAS20 response rates at week 102 were 80.7% and 76.9%. ASAS40 and ASAS partial remission were also similar between groups. ADA positivity rates were comparable (week 102: 23.3% vs 27.4%). Adverse events led to treatment discontinuation during the extension study in 3 (3.3%) and 4 (4.8%) patients, respectively.
Conclusions This is the first study to show that switching from RP to its biosimilar CT-P13 is possible without negative effects on safety or efficacy in patients with AS. In the maintenance group, CT-P13 was effective and well tolerated over 2 years of treatment.
Trial registration number NCT01571206; Results.
- Ankylosing Spondylitis
- Treatment
- Anti-TNF
- DMARDs (biologic)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
CT-P13 is a biosimilar of infliximab, a chimerical human-murine monoclonal antibody against tumour necrosis factor (TNF). Biosimilar drugs such as CT-P13 are generic versions of innovator biologics, or ‘reference products’ (RPs).1 Regulatory authorities in Europe, the USA and elsewhere have defined strict criteria for biosimilar approval. Although exact criteria vary in different countries or regions, it is usually necessary to show that a biosimilar is highly similar to its RP in physicochemical and biological terms. In addition, clinical studies must establish statistical equivalence in pharmacokinetics and efficacy, and comparability of safety.2–4
CT-P13 has been approved by the European Medicines Agency (EMA) for all indications held by the infliximab RP. Extensive testing has shown that all major physicochemical characteristics and in vitro biological activities of CT-P13 and RP, including affinity for soluble and transmembrane TNF, are highly comparable.5 ,6 CT-P13 and RP were also compared in two 54-week, multinational, randomised, double-blind, parallel-group studies in ankylosing spondylitis (AS) or rheumatoid arthritis (RA), namely Programme evaLuating the Autoimmune disease iNvEstigational drug cT-p13 in AS patients (PLANETAS) and Programme evaLuating the Autoimmune disease iNvEstigational drug cT-p13 in RA patients (PLANETRA). Analyses of these randomised studies established the pharmacokinetic and efficacy equivalence of CT-P13 and infliximab RP. Immunogenicity and safety findings for the two drugs were also similar.6–9
Due to the lower cost of biosimilars versus RPs, there is considerable interest in determining if newly diagnosed patients can be effectively and safely treated with a particular biosimilar, and whether those already on RP treatment can be switched to its biosimilar without adverse consequences on efficacy or safety.10 ,11 Collection of clinical data on this topic of ‘switchability’ is necessary. Here, we report the results of an open-label extension study that recruited patients with AS who had completed 54 weeks of the main PLANETAS study. This was a single-arm extension study in which all patients were pooled to be treated with CT-P13. The aims of the extension were twofold: (1) to investigate efficacy and safety during switched therapy to CT-P13 in patients previously treated with RP for 54 weeks in the main study (hereafter named the ‘switch group’); and (2) to investigate the efficacy and safety of CT-P13 treatment over 2 years in those patients treated with CT-P13 in the main study (the ‘maintenance group’). To aid interpretation, we describe the data for the maintenance and switch groups over two periods: the main study (week 0–54) and the extension study (week 54–102). A similar extension study that has recently been completed in patients with RA (PLANETRA) is also reported in this issue.12
Patients and methods
Methods of the main 54-week, randomised, double-blind, parallel-group PLANETAS study have been reported previously.6 ,7 Briefly, the main study recruited patients aged 18–75 years with active AS for ≥3 months according to the 1984 modified New York classification criteria. This Phase 1 PLANETAS extension study (ClinicalTrials.gov identifier: NCT01571206) was an open-label, single-arm extension study conducted in 40 centres in 8 countries. Patients who had completed 54 weeks of the main PLANETAS study were offered the opportunity of entering the extension study for another 1 year. Those who did not sign a new informed consent form for the extension were excluded. Additional eligibility criteria applied for the extension study were no major protocol violation in the main study and no new therapy for AS in the extension. Details of patient flow through the main and extension studies are shown in figure 1.
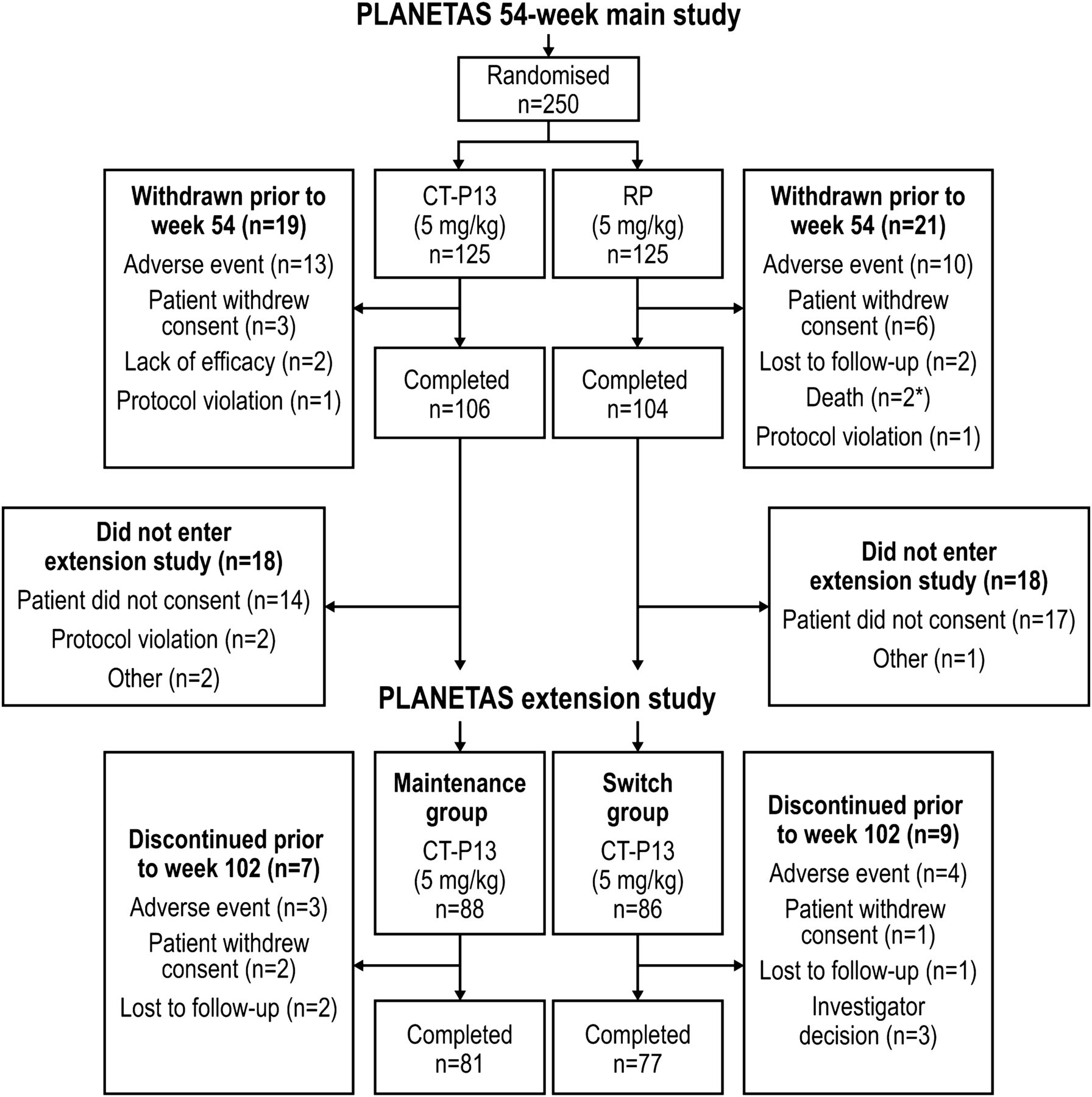
Patient disposition during the PLANETAS extension study. All patients who enrolled in the extension study (n=88 and n=86 in the maintenance and switch groups, respectively) were included in the ITT population. *One patient randomly assigned to RP received at least one dose of CT-P13 unintentionally. ITT, intent-to-treat; RP, reference product.
In the main PLANETAS study, patients received nine infusions of CT-P13 (CELLTRION, Incheon, Republic of Korea) or the infliximab RP (Janssen Biotech, Horsham, Pennsylvania, USA). After the ninth infusion of PLANETAS study treatment (given at week 54), eligible patients could choose to continue into the extension study during which patients and physicians were blinded to the treatment received in the main study. Patients in the extension study received an additional six infusions of CT-P13 given every 8 weeks from week 62 to week 102. CT-P13 was administered via 2 h intravenous infusion at a dose of 5 mg/kg. At the discretion of the investigator, antihistamines could be provided 30–60 min before CT-P13 infusion. The extension study was performed according to the principles of the Declaration of Helsinki and International Conference on Harmonisation good clinical practice guidelines. The relevant independent ethics committees approved the study protocol, consent forms and other written information for the extension study.
Study end points
Efficacy assessments were made at baseline and weeks 14, 30, 54, 78 and 102. Efficacy end points included the proportion of patients achieving clinical response according to Assessment of SpondyloArthritis international Society (ASAS)20 and ASAS40 criteria; the proportion of patients with ASAS partial remission (PR); the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), the Bath Ankylosing Spondylitis Functional Index, the Bath Ankylosing Spondylitis Metrology Index, and the Ankylosing Spondylitis Disease Activity Score (ASDAS)-C reactive protein (CRP).
For assessment of immunogenicity, the proportion of patients with antidrug antibodies (ADAs) was assessed at baseline and weeks 14, 30, 54, 78 and 102 using an electrochemiluminescent immunoassay method, as previously reported.6 ,7 The neutralising activity of ADAs was also assessed by a flow-through immunoassay method using the Gyros Immunoassay platform (Gyros AB, Sweden). For safety, treatment-emergent adverse events (TEAEs) were assessed throughout the main and extension studies. The following TEAEs of special interest were also monitored: infusion-related reactions (including hypersensitivity and anaphylactic reaction), tuberculosis (TB), latent TB (defined as a positive conversion on an interferon-γ release assay—which was negative at baseline—together with a negative result on chest X-ray examination), serious infection, pneumonia, drug-induced liver injury, vascular disorders and malignancies. Other safety assessments included monitoring of vital signs, physical examination findings and clinical laboratory analyses. Details of exploratory and post hoc end points are included in online supplementary appendix A.
Supplemental material
Statistical analyses
Statistical analyses were conducted using SAS software V.9.1.3 (SAS Institute, Cary, North Carolina, USA). Continuous data were summarised using descriptive statistics. Categorical data were summarised using counts and percentages. The populations were predefined in the study protocol and statistical analysis plan for participants in the extension study. The intent-to-treat (ITT) population consisted of all enrolled patients. The efficacy population consisted of all patients who received at least one full dose of study treatment and had data for at least one efficacy assessment in the extension study. All efficacy analyses were performed using a ‘missing equals excluded’ (MEX) approach. A logistic regression model was used to analyse the proportion of patients achieving clinical response (ASAS20/ASAS40/ASAS PR), with treatment group as a fixed effect and the stratification factors (baseline BASDAI score and region) as covariates. Treatment effect differences between maintenance and switch groups were estimated by calculating ORs and their 95% CI. Descriptive statistics were used to analyse other efficacy end points. The safety population (used to analyse all safety and immunogenicity events) consisted of all patients who received at least one dose of study treatment in the extension study. Data from the main study period were analysed in participants of the extension study only, and not in all patients from the main study. Methods for sensitivity analyses of ASAS responses are included in online supplementary appendix A.
Results
Patients
The first patient visit in the main PLANETAS study was held in November 2010; the last patient visit in the extension study took place in June 2013. A total of 174 of 210 patients from the main study who consented to participate in the extension study were screened under the approval of the appropriate ethics committee (figure 1). All 174 screened patients were enrolled and were included in the ITT, efficacy and safety populations of the extension study. Of these, 88 had been randomised to CT-P13 in the main study and 86 to RP. Thus, the ITT population for the extension study included 88 and 86 patients in the maintenance and switch groups, respectively. Patient demographics and baseline characteristics were broadly comparable between these two groups (table 1). In the ITT population, 81 (92.0%) and 77 (89.5%) patients in the maintenance and switch groups, respectively, completed all 48 weeks of the extension study; 7 (8.0%) and 9 (10.5%) patients discontinued the extension study. Reasons for discontinuation are detailed in figure 1. Two patients randomised to RP in the main study were incorrectly treated with CT-P13 during that study. Applying a conservative approach, those two patients were classified as being in the CT-P13 group for safety analyses in the main study. Therefore, 90 and 84 patients were included in the maintenance and switch groups, respectively, in the safety population of the extension study.
Patient demographics and baseline characteristics in the PLANETAS extension study (ITT population)
Efficacy
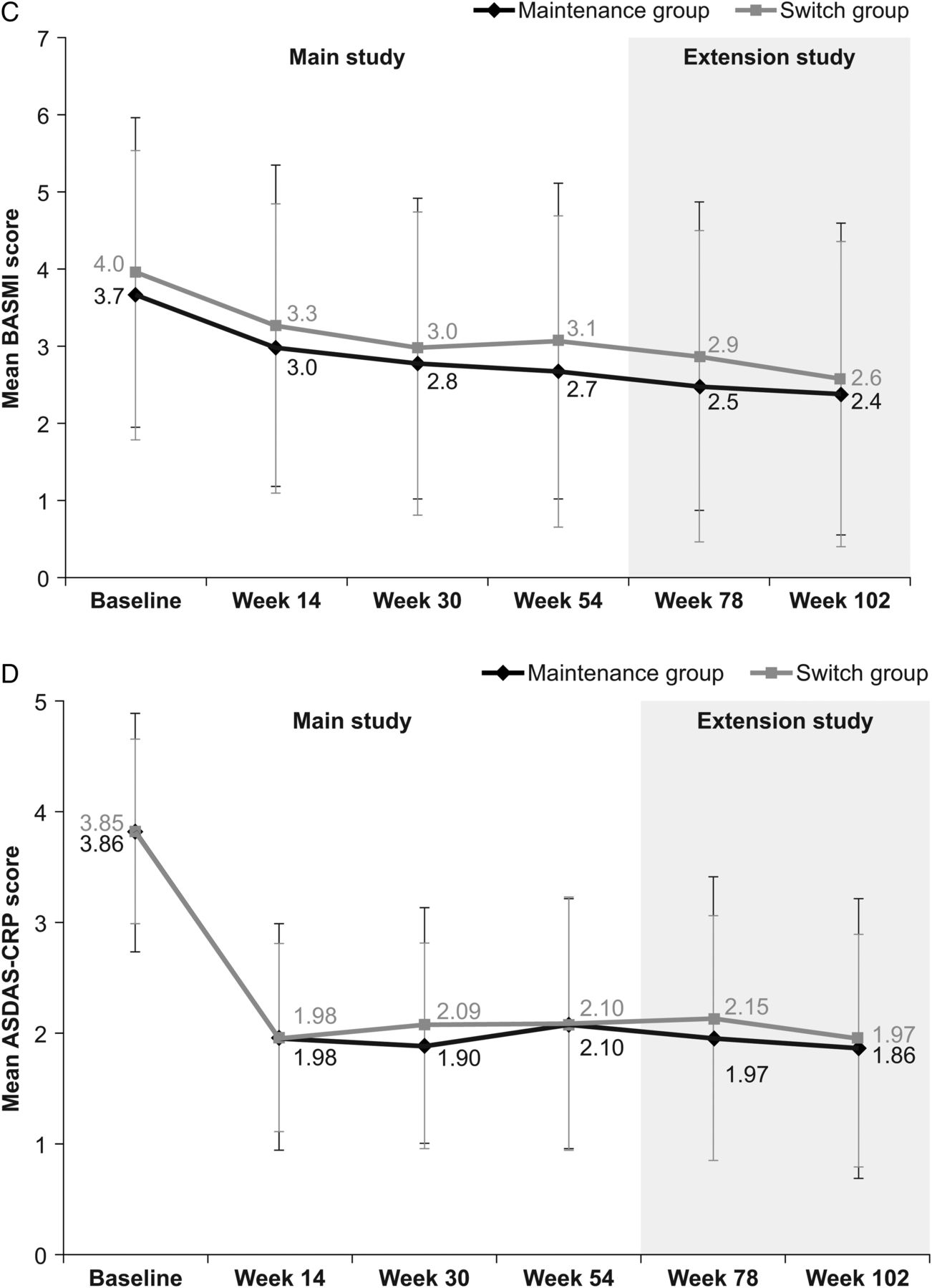
During the extension study (ie, week 54–102), the proportion of patients achieving a clinical response according to ASAS20 and ASAS40 criteria, and ASAS PR rates, were maintained at similar levels to those in the main study in both the maintenance and switch groups, and were comparable between groups (table 2). In the switch group, response rates at week 102 (ie, 48 weeks after the last infusion of RP at week 54) were similar to those observed before switching (ie, at week 54; ASAS20: 76.9% at week 102 vs 75.6% at week 54; ASAS40: 61.5% vs 53.5%; ASAS PR: 23.1% vs 17.4%). Response rates at week 102 were also similar to those observed at week 54 in the maintenance group (ASAS20: 80.7% at week 102 vs 70.5% at week 54, respectively; ASAS40: 63.9% vs 58.0%; ASAS PR: 19.3% vs 17.0%). Among patients who participated in the extension study, clinical response rates were also similar between groups throughout the main study (week 0–54) (table 2). In a subgroup analysis performed according to ADA status, the proportion of ADA-negative patients achieving ASAS20 was 75.7% at week 54 and 83.9% at week 102 in the maintenance group, and 79.0% at week 54 and 84.6% at week 102 in the switch group. In comparison, 50.0% (week 54) and 70.0% (week 102) of ADA-positive patients in the maintenance group, and 66.7% (week 54) and 62.5% (week 102) in the switch group achieved ASAS20 (online supplementary appendix B, figure B-1). As summarised in figure 2⇓, there were no noticeable changes in other efficacy end points during the PLANETAS extension, either between or within groups.
Proportion of patients with AS with an ASAS20 response, ASAS40 response and ASAS PR* (efficacy population in PLANETAS extension study with MEX approach)

Additional efficacy end points assessed in the PLANETAS extension study. Mean (SD) BASDAI (A), BASFI (B), BASMI (C) and ASDAS-CRP (D) scores in maintenance* (n=88) and switch** (n=86) groups during the main study and the extension study (efficacy population). *Patients treated with CT-P13 during the 54 weeks of the main study and the 48-week extension study. **Patients treated with RP during the 54 weeks of the main study and then switched to CT-P13 during the 48-week extension study. ASDAS, Ankylosing Spondylitis Disease Activity Score; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; BASMI, Bath Ankylosing Spondylitis Metrology Index; CRP, C reactive protein; RP, reference product.

{kind=link}
{kind=link}
{kind=link}
Continued.
Sensitivity analyses to compare populations and statistical approaches appeared to support sustained efficacy and comparability between the two groups. Analyses using the last observation carried forward (LOCF) approach showed highly similar results to those using the MEX approach, both in the maintenance group (ASAS20: 80.7% at week 102 with both LOCF and MEX approaches) and the switch group (ASAS20: 74.4% at week 102 using LOCF vs 76.9% using MEX). Analyses of the main study ITT population using the LOCF and non-responder imputation (NRI) approaches showed relatively low response rates compared with the extension study ITT population. However, when analysed using the LOCF approach, response rates were comparable between the two groups and were sustained throughout the 2-year study period, both in the extension study ITT population (ASAS20: 80.7% at week 102 vs 70.5% at week 54 in the maintenance group, 74.4% vs 75.6% in the switch group) and the main study ITT population (ASAS20: 65.6% at week 102 vs 58.4% at week 54 in the CT-P13 group, 64.8% vs 65.6% in the RP group) (online supplementary appendix C, tables C-1 and C-2). When analysed using the NRI approach, lower response rates were seen at week 102 than week 54 but rates were still comparable between the groups (ASAS20: 53.6% at week 102 vs 56.8% at week 54 in the maintenance group; 48.0% vs 60.0% in the switch group) (online supplementary appendix C, table C-3).
Immunogenicity
During the main and extension studies, the proportion of patients with ADAs was similar in the maintenance and switch groups at each time point (table 3). At week 54, the proportions of patients with ADAs in the maintenance and switch groups were 22.2% and 26.2%, respectively. In the extension study, these figures were 23.3% and 29.8% at week 78 and 23.3% and 27.4% at week 102. Between-group differences did not reach statistical significance at any time point. In the switch group, ADA incidence did not increase from week 54 to 102 (26.2% vs 27.4%). In general, most of the patients with a positive ADA result were also positive for neutralising antibodies. The proportion of ADA-positive patients with sustained ADAs was similar in the two groups (85.7% and 88.9% in the maintenance and switch groups, respectively). There was no analysis for IgG4.
Proportion of patients with ankylosing spondylitis (AS) who were positive for antidrug antibodies (ADAs) and neutralising antibodies (NAbs) in PLANETAS: the main 54-week parallel-group study and the extension study (safety population)
Pharmacodynamics
In a subgroup analysis by ADA status, mean change from baseline in CRP and erythrocyte sedimentation rate (ESR) was comparable in the maintenance and switch groups at week 54 and week 102 in both ADA-negative and ADA-positive patients (see online supplementary appendix B, table B-1).
Safety
The proportion of patients who experienced at least one TEAE was 48.9% (n=44 of 90) in the maintenance group and 71.4% (n=60 of 84) in the switch group during the extension study, and 70.0% (n=63) and 61.9% (n=52) during the main study. Online supplementary appendix D lists all TEAEs reported during the extension study (see online supplementary table D-1). Rates of TEAEs in both groups during the main and extension studies were within the range reported in historical studies with infliximab RP (online supplementary appendix E). TEAEs considered by the investigator to be related to study treatment were reported in 20 (22.2%) and 33 (39.3%) patients in the maintenance and switch groups, respectively, during the extension study, and in 40 (44.4%) and 35 (41.7%) patients during the main study. During the extension phase, reported treatment-related TEAEs included infusion-related reactions (7.8% and 7.1% in the maintenance and switch groups, respectively), abnormal liver function tests (4.4% and 4.8%), latent TB (2.2% and 4.8%) and upper respiratory tract infection (3.3% and 2.4%) (table 4). Most treatment-related TEAEs were mild to moderate in severity (45.6% (41 of 90) and 65.5% (55 of 84) in maintenance and switch groups, respectively).
Treatment-related TEAEs reported in at least 1% of patients in total, n (%) (safety population)
TEAEs led to discontinuation of treatment during the extension study in 3 (3.3%) and 4 (4.8%) patients in the maintenance and switch groups, respectively. In both groups, four patients experienced treatment-emergent serious adverse events (SAEs) during the extension study. Two of these SAEs were considered related to treatment (TB, one in each group). During the main study, four (4.4%) and two (2.4%) patients in the maintenance and switch groups, respectively, experienced treatment-emergent SAEs. One of these events, which occurred in a patient in the switch group, was considered to be related to treatment (cellulitis). All these SAEs were resolved except one SAE (prostate cancer) which was reported in the maintenance group during the extension study and which was considered to be unrelated to the study drug by the investigator.
Data on TEAEs of special interest are shown in table 5. Infusion-related reactions were reported in seven (7.8%) patients in the maintenance group and six (7.1%) in the switch group during the extension study. Of these, four patients in each group were ADA-positive (see online supplementary appendix B, table B-2). Among patients with infusion-related reactions, none experienced anaphylaxis. During the main study period, infusion-related reactions were reported in four (4.4%) and seven (8.3%) patients in the maintenance and switch groups, respectively. Of these, one and four patients, respectively, were ADA-positive. Latent TB occurred in five (5.6%) and seven (8.3%) patients in the maintenance and switch groups, respectively, during the extension study, and in six (6.7%) and three (3.6%) patients in the main study. Patients reporting latent TB received TB prophylaxis according to local guidelines. Among such patients, two reported active TB SAEs during the extension study (one in each group), as reported above. One malignancy was observed during the extension study (the case of prostate cancer described above).
TEAEs of special interest regardless of relationship to study treatment in the PLANETAS main and extension study, n (%) (safety population)
Discussion
The open-label extension study reported here evaluated the efficacy and safety of treatment with a maximum of six infusions of CT-P13 in patients with AS who had previously been treated with either CT-P13 (maintenance group) or RP (switch group) for 54 weeks. In the switch group, ASAS response rates were maintained from week 54 (ie, the time of the last RP infusion) to week 102 (ie, 48 weeks after the last RP infusion). In the maintenance arm, responses to CT-P13 observed in the preceding 54-week study were maintained throughout the open-label extension study. As previously reported,6 ,7 efficacy findings at weeks 30 and 54 of PLANETAS were comparable to 24-week and 54-week data from the infliximab arm of ASSERT (Ankylosing Spondylitis Study for the Evaluation of Recombinant infliximab Therapy) and another pivotal placebo-controlled study of RP in AS.13–15 Similarly, ASAS response rates in the maintenance group at the end of the PLANETAS extension (ie, week 102) were comparable to those observed at week 102 of ASSERT (ASAS20: 80.7% vs 73.9%, respectively; ASAS40: 63.9% vs 59.4%).16 Data for all other efficacy end points were also similar between the maintenance and switch groups throughout the main and extension study periods, as well as within each group over both study periods. Sensitivity analyses supported the findings of sustained efficacy and comparability between groups. LOCF and MEX approaches reported highly similar results, both in the maintenance and switch groups. Analyses of the main study ITT and extension study ITT populations showed comparable and sustained outcomes throughout the 2-year study. Although analysis of the main study ITT population using the NRI approach showed lower response rates at week 78 and week 102 than when the LOCF approach was used, response rates were still comparable between groups. Differences in response rates by analysis method were likely due to the fact that some patients who responded during the main study were not included in the extension (figure 1); therefore, the LOCF approach can be considered the most appropriate method.
In this extension study, CT-P13 was well tolerated, displaying a safety profile over the long term that was consistent with the profile of infliximab RP.16 ,17 No notable differences were observed between the maintenance and switch groups in the incidence of TEAEs leading to discontinuation from the study (3/90 and 4/84, respectively), SAEs (4 patients in either group), serious infections including active TB, ADAs or infusion reactions (tables 3 and 5). A lower proportion of patients in the maintenance group than the switch group experienced one or more TEAE during the extension. To further evaluate the numerical imbalance in the proportions of patients reporting TEAEs during the extension study, a safety meta-analysis using data from historical studies conducted with infliximab RP was performed. For this purpose, a literature search was conducted to identify studies in patients with AS focusing on randomised studies or those with an observational design, with a duration of at least 54 weeks and long-term cohorts, and observational studies capturing safety reporting up to 2 years. Only studies which reported the methodology of collecting safety data and incidence of adverse events using reliable denominators were included in the meta-analysis. The result of the meta-analysis is presented in online supplementary figure E-1. The proportion of patients reporting TEAEs in both the maintenance and switch groups was within the range reported in historical studies with infliximab RP.6 ,16 ,18–22 Of note, the majority of TEAEs in the switch group were generally mild to moderate in severity. In addition, the incidence of infusion-related reactions did not increase in patients who switched from RP to CT-P13, while the proportion of patients with sustained or transient ADAs was comparable between groups throughout the study, although only qualitative analyses of these data were performed. In a similarly designed extension study of PLANETRA—a Phase III study which compared CT-P13 and RP in patients with RA —the incidence of TEAEs in the maintenance and switch groups was similar (see online supplementary appendix F).12 In this regard, there was no indication of a change in the safety profile when patients transitioned from infliximab RP to CT-P13.
It is well known that development of ADAs against infliximab is associated with a decreased clinical response and is also linked to adverse effects such as infusion-related reactions.23 ,24 We observed lower ASAS20 response rates, higher levels of CRP and ESR, and a higher incidence of infusion-related reactions in ADA-positive compared with ADA-negative patients. These effects were comparable in both groups, suggesting that switching from RP to CT-P13 did not have a negative impact, regardless of ADA status.
The current data suggest that switching to CT-P13 in patients previously treated with the infliximab RP shows similar efficacy and safety profiles, compared with maintaining CT-P13. A number of randomised and non-randomised studies have investigated the issue of switchability for other biosimilar drugs, including those such as the epoetins, granulocyte-colony stimulating factors and human growth hormone that have been approved in Europe for several years.25–31 In general, these studies suggest there are unlikely to be adverse consequences of switching between innovator biologics and those biosimilars that have undergone the rigorous level of scrutiny necessary for approval by the EMA. The similarly designed extension of the PLANETRA study in patients with RA also showed no adverse effects of switching to CT-P13 from RP.12
It is important to note that this single-arm, open-label extension study was neither designed nor powered to formally evaluate the non-inferiority or equivalence of switching from RP to CT-P13 vs continued treatment with CT-P13. In this regard, a randomised, double-blind, Phase IV study has been initiated in Norway (‘NOR-SWITCH’; ClinicalTrials.gov identifier: NCT02148640). NOR-SWITCH is comparing the safety and efficacy of switching from the infliximab RP to CT-P13 vs continued treatment with RP in adults with RA, spondyloarthritis, chronic psoriasis, psoriatic arthritis, ulcerative colitis or Crohn's disease. In addition, a pharmacovigilance programme has been initiated by the manufacturers of CT-P13. This programme will monitor the safety of CT-P13 in patients with various inflammatory diseases, including AS, who have switched from RP, and who are receiving infliximab treatment for the first time.
Conclusions
CT-P13 was well tolerated with comparable efficacy and safety as the historical infliximab RP treatment in patients with AS over 102 weeks. Furthermore, switching from the infliximab RP to CT-P13 after 1 year of infliximab RP treatment showed continued comparable efficacy, immunogenicity and safety, to maintenance of CT-P13 treatment during the 2nd year of the treatment.
Acknowledgments
The authors thank the patients and study personnel who made this trial possible, and the PLANETAS study investigators: Bulgaria: Rashkov R; Chile: Goecke Sariego I, Gutierrez M; Korea (Republic of): Kang SW, Kim HY, Kim TH, Park YB, Park YE, Song JS, Suh CH; Latvia: Andersone D, Saulite-Kandevica D; Mexico: Araiza R, Morales-Torres J, Pacheco-Tena C; Poland: Brzezicki J, Jaworski J, Korkosz M, Krogulec M, Piotrowski M, Jeka S, Hrycaj P; Spain: Blanco Garcia F, Díaz-González F; Ukraine: Amosova K, Kovalenko V, Dyadyk O, Gnylorybov A, Ter-Vartanian S; USA: Ahn C. The authors thank Rick Flemming (Aspire Scientific Limited, Bollington, UK) for editorial support (writing assistance, assembling tables and figures, collating author comments, grammatical editing and referencing). Editorial support was funded by CELLTRION Healthcare Co., Ltd (Incheon, Republic of Korea).
References
Footnotes
Handling editor Tore K Kvien
Contributors WP, DHY, JB, SJL, HUK, YJB and SYK were involved in the conception and design of the study, and/or analysis and interpretation of data, drafting of the manuscript and revising it critically for important intellectual content, and final approval of the version to be published. PM, MB, PW, SG-U, HM, Y-AL, SS, M-JL, VK and CA-M were involved in the acquisition of data, drafting of the manuscript and revising it critically for important intellectual content, and the final approval of the version to be published.
Funding This study was funded by CELLTRION Inc (Incheon, Republic of Korea).
Disclaimer The sponsor participated in study design, in the collection, analysis and interpretation of study data, and in reviewing drafts of the manuscript. The final decision to submit the manuscript was made by the authors.
Competing interests DHY and WP: Consultation for Celltrion. MB has received research grants from Celltrion, personal fees from lectures for Roche, Abbvie, MSD, Pfizer, Egis, UCB outside the submitted work; JB has received honoraria for talks, advisory boards, paid consultancies or grants for studies from Abbvie (Abbott), Amgen, Boehringer Ingelheim, Bristol-Myers-Squibb, Celgene, Celltrion, Centocor, Chugai, EBEWE Pharma, Janssen, Medac, MSD (Schering-Plough), Mundipharma, Novartis, Pfizer (Wyeth), Roche, Sanofi-Aventis and UCB. HUK, SJL, YJB and SYK are full-time employees of Celltrion. Otherwise, none declared.
Patient consent Obtained.
Ethics approval The protocol was reviewed and approved by each site's institutional review board or independent ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data available for this paper are included in the manuscript and online supplementary appendices.