Article Text

Abstract
Objective To evaluate apremilast treatment in patients with active psoriatic arthritis, including current skin involvement, despite prior therapy with conventional disease-modifying antirheumatic drugs and/or biologic agents.
Methods Patients (N=505) were randomised (1:1:1) to placebo, apremilast 20 mg twice daily, or apremilast 30 mg twice daily. Rescue therapy with apremilast was designated at week 16 for placebo patients not achieving 20% improvement in swollen and tender joint counts. At week 24, the remaining placebo patients were then randomised to apremilast 20 mg twice daily or 30 mg twice daily. The efficacy and safety of apremilast were assessed over 52 weeks.
Results At week 16, significantly more patients receiving apremilast 20 mg twice daily (28%) and 30 mg twice daily (41%) achieved 20% improvement in American College of Rheumatology response criteria versus placebo (18%; p=0.0295 and p<0.0001, respectively), and mean decrease in the Health Assessment Questionnaire-Disability Index score was significantly greater with apremilast 30 mg twice daily (−0.20) versus placebo (−0.07; p=0.0073). In patients with baseline psoriasis body surface area involvement ≥3%, significantly more apremilast 30 mg twice daily patients achieved 50% reduction from baseline Psoriasis Area and Severity Index score (41%) versus placebo (24%; p=0.0098) at week 16. At week 52, observed improvements in these measures demonstrated sustained response with continued apremilast treatment. Most adverse events were mild to moderate in severity; the most common were diarrhoea, nausea, headache and upper respiratory tract infection.
Conclusions Apremilast demonstrated clinically meaningful improvements in psoriatic arthritis and psoriasis at week 16; sustained improvements were seen with continued treatment through 52 weeks. Apremilast was generally well tolerated and demonstrated an acceptable safety profile.
Trial registration number NCT01212770.
- Psoriatic Arthritis
- Ankylosing Spondylitis
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) is a chronic immune disease. Manifestations of PsA include swollen and tender joints, pain, and enthesitis and dacylitis, which are associated with impaired physical function and health-related quality of life.1–4 There is little evidence of effect for conventional disease-modifying antirheumatic drugs (DMARDs), such as methotrexate, in PsA treatment.5–9 When assessing the effectiveness of a therapy for PsA, it is important to understand efficacy in the context of active skin disease and whether the therapy improves features specifically associated with PsA.
Apremilast inhibits phosphodiesterase 4 (PDE4) conversion of cyclic AMP to AMP, thus indirectly downregulating the inflammatory response through decreased inflammatory cytokine expression and increased expression of anti-inflammatory cytokines.10 The effect of apremilast on multiple manifestations of PsA is being assessed in patients with active PsA in the Psoriatic Arthritis Long-term Assessment of Clinical Efficacy (PALACE) phase III clinical trial programme. Previously reported results of PALACE 1 have demonstrated the efficacy of apremilast in the treatment of PsA.11 ,12
PALACE 3 assessed apremilast treatment across different aspects of PsA in patients with active disease, including current skin disease. This report describes the results of the first 52 weeks of PALACE 3.
Methods
Patients
The study enrolled adults with PsA of ≥6 months’ duration who met the Classification Criteria for Psoriatic Arthritis (CASPAR) at screening and had ≥three swollen and ≥three tender joints. All patients were required to have had prior treatment with conventional DMARDs and/or biologics; tumour necrosis factor inhibitor efficacy failures were limited to ≤10% of randomised patients. Patients were also required to have active skin disease with ≥one plaque psoriasis skin lesion ≥2 cm in size.
Patients were excluded if they had prior therapeutic failure of >three DMARDs or >one tumour necrosis factor blocker; history of or current rheumatic disease or autoimmune joint disease other than PsA or functional class IV status as defined by the American College of Rheumatology (ACR) Classification of Functional Status in Rheumatoid Arthritis; erythrodermic, guttate or generalised pustular psoriasis; malignancy (except treated basal or squamous cell skin carcinoma or early forms of cervical carcinoma with no recurrence in 5 years); prior treatment with apremilast; or phototherapy (ultraviolet B, psoralen+ultraviolet A) or immunosuppressive systemic therapy (except as noted) within 4 weeks, adalimumab, etanercept, golimumab, infliximab, certolizumab pegol, or tocilizumab within 12 weeks, or alefacept or ustekinumab within 24 weeks of randomisation. Patients with active tuberculosis, history of incompletely treated tuberculosis or significant infection within 4 weeks of screening were excluded. No purified protein derivative or QuantiFERON screening for latent tuberculosis was required. There was no protocol requirement to interrupt study medication for patients who developed infection during the study, and no protocol prohibition on vaccinations.
Patients taking concurrent DMARDs could continue stable doses (≥4 weeks before baseline) of methotrexate (≤25 mg/week), leflunomide (≤20 mg/day), sulfasalazine (≤2 g/day), or combination. One DMARD dose reduction was allowed after week 24. Stable doses of oral corticosteroids (prednisone ≤10 mg/day or equivalent for ≥4 weeks before baseline) and non-steroidal anti-inflammatory drugs (for ≥2 weeks before baseline) were permitted. Low potency topical corticosteroids for treatment of face, axillae and groin psoriatic lesions, coal tar shampoo and/or salicylic acid scalp preparations for scalp lesions, and non-medicated emollient for body lesions were permitted, except ≤24 h before study visits. The use of DMARDs other than methotrexate, leflunomide or sulfasalazine was not permitted ≤4 weeks of randomisation. Except for treatments permitted as described above, topical therapy for psoriasis ≤2 weeks of randomisation was prohibited.
All patients provided written informed consent.
Study design
PALACE 3 is a phase III, randomised, placebo-controlled study (see online supplementary figure S1). In the placebo-controlled, double-blind phase, patients were randomised (1:1:1) to placebo, apremilast 20 mg twice daily, or apremilast 30 mg twice daily using a centralised interactive voice response system. Patients were stratified by baseline DMARD use (yes/no) and psoriasis involvement of the body surface area (BSA; <3%/≥3%). Apremilast was dose-titrated over the 1st week of treatment (10 mg on day 1, with increases of 10 mg/day until the target dose was reached). Patients whose swollen joint count (SJC) and tender joint count (TJC) had not improved by ≥20% at week 16 were considered non-responders and were then randomised (1:1) to apremilast 20 mg twice daily or 30 mg twice daily if initially randomised to placebo, or continued on their initial apremilast dose. At week 24, all remaining placebo patients were then randomised to apremilast 20 mg twice daily or 30 mg twice daily. Patients continued in a 28-week double-blind, active treatment period up to week 52. A long-term extension phase is ongoing.
Efficacy assessments
The primary efficacy end point was the proportion of patients achieving an ACR20 response at week 16 modified for PsA by the addition of distal interphalangeal joints of the toes and the carpometacarpal joints to TJC and SJC.13 ,14 The key secondary end point was the change from baseline in the Health Assessment Questionnaire-Disability Index (HAQ-DI) at week 16.15 Additional efficacy measures included improvements in SJC and TJC; physical function; enthesitis, measured by the presence or absence of pain at 13 select entheses and tendon insertions16; dactylitis, based on the number of digits on the hands and feet with dactylitis present; and psoriasis activity, based on the proportion of patients evaluable by the Psoriasis Area and Severity Index (PASI) (baseline psoriasis involvement ≥3% BSA) who achieved ≥75% or ≥50% reductions from baseline (PASI-75 or PASI-50, respectively). Efficacy end points were assessed at week 16, week 24 and week 52; ACR20 response, HAQ-DI scores, and SJC and TJC were also evaluated at week 40.
Safety assessments
Safety assessments were conducted at screening, week 0, week 4, week 16, week 24, week 28, week 40 and week 52, and in the event of early termination. Assessments included collection of adverse events (AEs), clinical laboratory testing, physical examinations, and vital signs at each visit, and 12-lead ECG at screening and week 0, week 16, week 24 and week 52.
Statistical analysis
Sample size estimations were based on a phase II study of apremilast,17 which showed that a sample of 165 patients per group would be needed to achieve 95% power to detect a 20% absolute difference in the ACR20 response between apremilast treatment and placebo using a two-group χ2 test at a two-sided significance level of 0.025.
Efficacy at week 16 was evaluated for the intent-to-treat population, which included all randomised patients who received ≥one dose of study medication. Missing values were handled using the non-responder imputation for categorical variables and last-observation-carried-forward (LOCF) approach for continuous variables. ACR20 response rates and other categorical variables at week 16 were compared using the Cochran-Mantel-Haenszel test, controlling for baseline DMARD use and baseline psoriasis (BSA <3%/≥3%). Changes in HAQ-DI and other continuous variables from baseline to week 16 were compared using an analysis of covariance (ANCOVA) model with treatment, baseline DMARD use (yes/no), and baseline psoriasis (BSA <3%/≥3) as factors and baseline value as a covariate; per cent change from baseline was analysed based on the ANCOVA model using rank transformation.
For the assessment of changes in enthesitis and dactylitis scores at week 24, values for all patients who qualified for early escape at week 16 were defined as missing at week 24 and imputed using LOCF for the week 24 analysis.
Efficacy analyses with no comparisons to placebo, including those at week 52, were performed using the observed data for each time point, which was the prespecified analysis at week 52.
Safety outcomes were assessed by study period and were analysed using the safety population, which comprised all patients who received ≥one dose of study medication. Safety data were analysed for two periods: the placebo-controlled period of week 0 to week 24 (data through week 16 for patients who initially received placebo and escaped early; data through week 24 for all other placebo patients and for patients randomised to apremilast) and the 0-week to 52-week apremilast-exposure period (data are included for all patients who received ≥one dose of apremilast, regardless of when treatment started (week 0, week 16 or week 24)). AEs were classified using the Medical Dictionary for Regulatory Activities classification system. Events occurring after the first dose of study medication and ≤28 days after the last dose were summarised descriptively.
Results
Of 612 patients screened, 505 were randomised and received ≥one dose of study medication and were included in the intent-to-treat population (figure 1). Of these, 469 (93%) completed week 16, with comparable completion rates among apremilast and placebo groups (92–93%). Overall completion rates for week 0–52 in apremilast 20 mg twice daily and 30 mg twice daily patients were 71% (120/169) and 75% (126/167), respectively. In addition, 72% (122/169) of patients randomised to placebo at baseline completed week 52.

Patient disposition through week 52. *Includes withdrawal by patient, lost to follow-up, protocol violation, non-compliance, other. †Includes patients initially randomised to placebo, who met early escape criteria and were then randomised to apremilast at week 16. ‡Includes patients initially randomised to placebo who were then randomised to apremilast at week 24 for the active treatment phase (week 24–52). AE, adverse event.
Randomised patients were representative of a population with active PsA and were comparable across treatment arms for baseline demographic and disease characteristics, as well as prior and concurrent PsA-related therapy (table 1).
Baseline demographic and clinical characteristics: intent-to-treat population (N=505)*
Efficacy
At week 16, significantly more apremilast 20 mg and 30 mg patients achieved an ACR20 response versus placebo (placebo: 18%; 20 mg: 28%, p=0.0295; 30 mg: 41%, p<0.0001) (figure 2A). The adjusted differences in response rate (95% CI) versus placebo were 10% (1.1% to 18.6%) with apremilast 20 mg and 22% (13.0% to 31.6%) with 30 mg.

ACR20 (A), HAQ-DI (B), SJC and TJC (C) and PASI-50 (D) at week 16. *p<0.05; †p≤0.0001 versus placebo, based on analysis of covariance model for HAQ-DI, SJC and TJC, and Cochran-Mantel-Haenszel test for ACR20 and PASI-50 for the intent-to-treat population; patients who discontinued or did not have sufficient data were counted as non-responders (ACR20 and PASI-50) or had their last observation was carried forward (HAQ-DI, SJC and TJC). Error bars represent SE. ACR, American College of Rheumatology; BSA, body surface area; HAQ-DI, Health Assessment Questionnaire-Disability Index; PASI-50, 50% reduction from baseline Psoriasis Area and Severity Index; SJC, swollen joint count; TJC, tender joint count.
In subgroup analyses (prior or concomitant treatment) at week 16, a consistent trend of higher ACR20 response rates in either apremilast dose group versus the placebo group was observed with and without concomitant conventional DMARD use and regardless of prior biologic use or failure. Biologic-naïve patients demonstrated numerically higher responses (see online supplementary table S1).
Apremilast 30 mg was associated with a significant improvement in the HAQ-DI score versus placebo at week 16 (figure 2B). Mean change from baseline in the HAQ-DI score at week 16 was −0.20 with apremilast 30 mg versus −0.07 with placebo (p=0.0073). Mean change in the HAQ-DI score with apremilast 20 mg (−0.13) did not reach statistical significance versus placebo. HAQ-DI improvement of ≥0.3018 was achieved by 31% of patients receiving apremilast 20 mg, 32% receiving apremilast 30 mg and 24% receiving placebo; differences did not reach significance at week 16.
Significantly more patients receiving apremilast 30 mg (41%; p=0.0098) achieved PASI-50 (post hoc analysis) versus placebo (24%) at week 16; PASI-50 achievement among patients receiving apremilast 20 mg (33%) was numerically higher but did not reach statistical significance (figure 2D). At week 16, significantly more apremilast 20 mg and 30 mg patients achieved PASI-75 versus placebo (placebo: 8%; 30 mg: 21%, p=0.0098; 20 mg: 20%, p=0.0215) (table 2).
Efficacy end points at week 16 (intent-to-treat population) and week 52 (data as observed)
Apremilast 30 mg was associated with significant results versus placebo at week 16 in other prespecified end points, including decreases in 28-joint disease activity score (DAS-28) and the patient's and physician's global assessments of disease activity at week 16. Significant improvements in mean and mean per cent change in TJC at week 16 were seen with both apremilast doses. Significant changes versus placebo at week 16 were not observed with either dose of apremilast in proportions of patients attaining ACR50 or ACR70, the mean change in C reactive protein (CRP), and despite numerical trend, mean per cent change in SJC; however, mean change in SJC was significant for apremilast 30 mg versus placebo (−3.5 vs −1.3, p=0.01; table 2).
Assessment of changes in enthesitis and dactylitis scores was preplanned at week 24 as a pooled analysis for the PALACE 1, 2 and 3 programme. We present here the PALACE 3 results at week 24. In patients with enthesitis at baseline, numerically greater improvements in the Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) were seen, with mean changes of −0.9 for apremilast 20 mg, −1.1 for apremilast 30 mg and −0.7 for placebo; results did not reach statistical significance.
In patients with dactylitis at baseline, mean change in dactylitis score at week 24 was significantly improved for apremilast 30 mg (−2.4, p=0.0399 vs placebo). Results for apremilast 20 mg did not reach statistical significance (−1.6) versus placebo (−1.4).
Sustained therapeutic effect of apremilast was observed across measures of efficacy through week 52 in patients continuing therapy (figure 3A–E and table 2).

{kind=link}
{kind=link}
{kind=link}
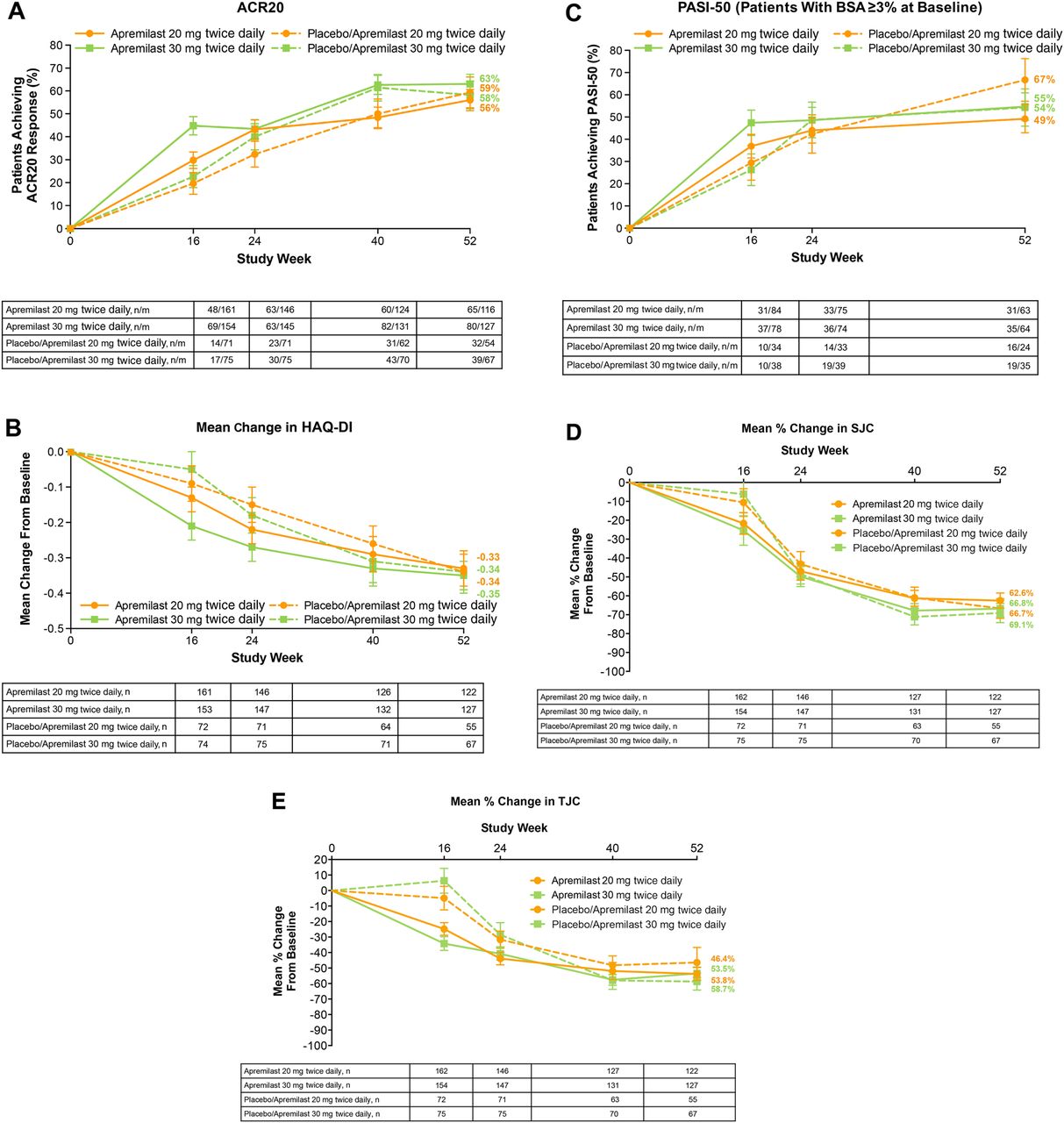
Over 52 weeks, ACR20 (A), HAQ-DI (B), PASI-50 (C), SJC (D) and TJC (E) (data as observed). Error bars represent SE. ACR, American College of Rheumatology; BSA, body surface area; HAQ-DI, Health Assessment Questionnaire-Disability Index; n/m=number of responders/number of patients with sufficient data for evaluation; PASI-50, 50% reduction from baseline Psoriasis Area and Severity Index score; SJC, swollen joint count; TJC, tender joint count.
Analysis of efficacy with longer-term treatment suggested sustained ACR20 response among patients initially randomised to apremilast and completing 52 weeks of study treatment. At week 52, 56% of patients receiving apremilast 20 mg and 63% receiving apremilast 30 mg achieved an ACR20 response. Sustained response rates were confirmed using an LOCF analysis at week 52 for any missing data; 52% of apremilast 30 mg patients achieved an ACR20 response (see online supplementary table S2). Similarly, changes in the HAQ-DI score and PASI-50 response in patients initially randomised to apremilast and completing 52 weeks of study treatment suggested sustained improvement in physical function and skin involvement. Mean changes in the HAQ-DI score were −0.33 (apremilast 20 mg) and −0.35 (apremilast 30 mg). Similar observations were seen with other secondary end points (table 2). Mean percentage improvements in SJC of −62.6% for apremilast 20 mg and −66.8% for apremilast 30 mg, as well as mean percentage improvements in TJC of −53.8% (apremilast 20 mg) and −53.5% (apremilast 30 mg) among patients treated for 52 weeks with apremilast indicates continued response to apremilast, as do mean changes in enthesitis (apremilast 20 mg: −2.2; apremilast 30 mg: −1.9) and dactylitis (apremilast 20 mg: −2.9; apremilast 30 mg: −3.6). Similarly, of patients who continued on apremilast 20 mg and 30 mg, 49% and 55% achieved PASI-50 and 29% and 39% achieved PASI-75, respectively. Treatment effects observed in patients initially randomised to placebo and then randomised to apremilast at week 16 or week 24 were consistent with patients initially randomised to apremilast (table 2).
Safety
Table 3 provides an overview of AEs during the 24-week placebo-controlled and 52-week apremilast-exposure phases. During the placebo-controlled phase (week 0–24), the most common AEs (≥5% of any treatment group) were diarrhoea, nausea, headache and upper respiratory tract infection (table 3). The majority of AEs were mild or moderate in severity. Discontinuations due to AEs during the 24-week placebo-controlled phase were ≤8% in any treatment group; few patients who received apremilast from baseline (n=7) discontinued due to AEs during week 24–52. Eighteen patients experienced a serious AE (SAE) during the placebo-controlled phase and 23 experienced an SAE during the 52-week apremilast-exposure phase. All SAEs during the apremilast-exposure phase were reported in one patient per treatment group, except worsening of PsA, which was reported in two apremilast 20 mg patients (table 3). No major adverse cardiac events occurred during the placebo-controlled or apremilast-exposure phases. Two malignancies (one breast cancer, one small cell lung cancer) were detected during the placebo-controlled period (both with apremilast 20 mg) and were not considered treatment related.
AEs and laboratory abnormalities during the placebo-controlled phase (week 0–24) and apremilast-exposure phase (week 0–52)
In the apremilast treatment groups, diarrhoea and nausea occurred at the highest frequency during the first 2 weeks of dosing, and decreased thereafter. Most AEs of diarrhoea and nausea were mild to moderate in severity and resolved within 30 days of onset with continued therapy and without medicinal intervention. In patients receiving apremilast from baseline and remaining on apremilast ≤52 weeks, new reports of diarrhoea and nausea were uncommon between week 24 and week 52 for apremilast 20 mg (diarrhoea: n=3/170; nausea: n=3/170) and 30 mg (diarrhoea: n=2/167; nausea: n=5/167). No SAEs of diarrhoea or nausea were reported throughout the 52-week apremilast-exposure phase. Overall, <2% of patients receiving apremilast discontinued because of diarrhoea or nausea through 52 weeks.
Marked abnormalities in vital signs, haematology and clinical chemistry variables were infrequent and occurred at a similar frequency with placebo or apremilast (table 3). Weight decrease was reported as an AE in a small proportion of patients during the 24-week placebo-controlled phase as well as during the 52-week apremilast-exposure phase. By week 24, mean change in weight from baseline to the last value measured was −0.05 kg (placebo), −1.2 kg (20 mg) and −1.2 kg (30 mg). Most patients maintained their weight ≤5% of baseline; at the end of the 52-week apremilast-exposure phase, weight loss >5% was observed in 14% (34/237) of apremilast 20 mg patients and 16% (38/237) of 30 mg patients.
Around 60% of participants were taking conventional DMARDs at the start of the trial (50% methotrexate with a mean dose of 14.75 mg/week). Common AEs were generally similar regardless of concomitant conventional DMARD use (see online supplementary table S3); no additional safety signals were observed.
Discussion
PsA is a chronic disorder that frequently requires long-term treatment. Because the pathophysiological mechanisms leading to psoriasis and PsA are closely tied, and most patients with PsA have coexistent psoriasis, it is important that agents used to treat PsA also demonstrate efficacy in improving psoriasis skin lesions. Patients enrolled in PALACE 3 had active PsA as well as active skin disease. This study demonstrated a significant therapeutic effect of apremilast 20 mg twice daily and 30 mg twice daily on disease activity, including improvement in signs and symptoms of PsA as well as psoriasis in a population with broad prior treatment experience.
Apremilast demonstrated clinically meaningful improvements in PsA disease severity at week 16, marked by significantly greater achievement of ACR20 response in both apremilast dose groups (vs placebo) as well as improvements in physical function based on the HAQ-DI (apremilast 30 mg), mean and mean per cent change in TJC, mean change in SJC (apremilast 30 mg), mean change in DAS-28 (CRP), patients achieving DAS-28 (CRP) <2.6, and physician's and patient's global assessments of disease activity (apremilast 30 mg); mean change in dactylitis count was significantly greater with apremilast 30 mg than with placebo at week 24. Despite numerical trends in some, ACR50/ACR70 responses, achievement of HAQ-DI minimal clinically important difference ≥0.30, change in CRP and mean per cent change in SJC were not significant at week 16. Similarly, week 24 changes in MASES were numerically greater with apremilast 30 mg, but not significant. A trend of higher ACR20 response rates with either apremilast dose (vs placebo), regardless of prior biologic exposure (biologic-naïve, biologic-experienced and biologic therapeutic failures) was seen, but responses were numerically lower for the biologic-experienced patients.
Psoriasis severity was significantly decreased at week 16 among patients with psoriasis BSA involvement of ≥3% at baseline. Although the study inclusion criteria mandated active skin disease in all patients, those evaluable by PASI constituted about 55% of the population enrolled, with PASI scores slightly lower than in PALACE 1.11 Efficacy data in patients with more severe psoriasis are available from the Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis (ESTEEM) studies.19 ,20 PASI responses in PsA are often not as robust as responses shown in psoriasis studies. Psoriasis involvement of <10% of BSA at baseline or baseline PASI scores <10 have been associated with poorer PASI responsiveness than that seen for patients with greater psoriasis involvement.21 ,22
Improvements in one or both apremilast dose groups (vs placebo) at week 16 in secondary end points, including health-related quality of life and measures of global PsA disease activity, demonstrated the shorter-term effect of apremilast across the manifestations of PsA. In patients remaining on apremilast therapy for ≤52 weeks, observed improvements from baseline across many of these measures suggest sustained response.
The observed safety profile was similar to that in previous apremilast investigations and consistent over the 24-week and 52-week phases. The most common AEs, diarrhoea and nausea, occurred most often during the first 2 weeks of apremilast treatment, with a reduced incidence after the 1st month of dosing, and were generally mild or moderate in severity. Laboratory abnormalities were infrequent and did not indicate a need for laboratory monitoring.
Efficacy and safety assessments during the active treatment phase describe the effects of apremilast observed among patients who continued its use over 52 weeks. The 52-week continuation rate in PALACE 3 was 73%, which is within the same range as the continuation rates observed in studies of biologics in patients with PsA.23–25 Long-term assessments may be biased based on the likelihood that patients not responding to or tolerating treatment may be more likely to discontinue. It is notable, however, that, over 52 weeks, <10% of patients discontinued apremilast because of AEs, and <10% discontinued because of lack of efficacy. A limitation of the PALACE study design includes the lack of radiographic assessment; further studies are needed to fully evaluate the effect of apremilast on joint damage progression. The short length of the placebo-controlled part of the study is also a limitation, as a meaningful analysis of safety is not possible in the context of a brief placebo-controlled phase (data through week 16 for patients who initially received placebo and escaped early; data through week 24 for all other patients). Whereas a lengthier placebo-controlled phase would provide more rigorous comparative data, it also presents ethical limitations; the week 24–52 active treatment period provided an alternative means to describe tolerability and safety of apremilast among those patients with longer-term exposure. Results from open-label extensions of the PALACE programme up to an additional 4 years will provide more information on the long-term use of apremilast.
These findings are important, as a need remains for long-term treatment options for patients with PsA, including those with active psoriasis skin lesions. Based on the significant efficacy seen with apremilast 30 mg in PALACE 3 in several domains of PsA, including ACR20 response, changes in HAQ-DI, mean and mean per cent change in TJC, mean change in SJC, dactylitis count, and DAS-28 (CRP), achievement of DAS-28 (CRP) <2.6, physician's and patient's global assessments of disease activity, and skin involvement, as well as a favourable risk:benefit profile and apparent lack of need for ongoing laboratory monitoring, apremilast represents a new oral treatment option, with a unique mode of action, for patients with PsA.
Acknowledgments
The authors received editorial support in the preparation of this report from Jennifer Schwinn, RPh, and Kristin Carlin, RPh, MBA, of Peloton Advantage, LLC, funded by Celgene Corporation. The authors, however, directed and are fully responsible for all content and editorial decisions for this manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Correction notice This article has been corrected since it was published Online First. At the end of the first paragraph of the Methods/Patients section >2 cm has been corrected to ≥2 cm.
Contributors All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. CJE had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study conception and design: FJB and JC. Acquisition of data: CJE, JC, CAB, JJ, JA and PB. Analysis and interpretation of data: CJE, FJB, JC, CAB, JJ, JA, RMS, AV, XZ and PB.
Funding This study was sponsored by Celgene Corporation.
Competing interests CJE has received research grants and consultant fees from Celgene, Pfizer, Roche and Samsung, and has served on the speaker's bureau for Abbott, Glaxo-SmithKline, Pfizer, Bristol-Myers Squibb, Janssen, Novo Nordisk, UCB and Roche. FJB has received consulting fees from Bioiberica, Gebro Pharma and Pfizer. JC has received research grants from AbbVie, Amgen, Celgene, Janssen, Merck and Pfizer; has received consulting fees from AbbVie and Amgen; and has served on the speaker's bureau for AbbVie. CAB has received research grants from Amgen, Bristol-Myers Squibb, Incyte, Eli Lilly, Merck and Pfizer. JA has received research grants and consultant fees from Ardea, Astra Zeneca, Bristol-Myers Squibb, Celgene, Centocor, Galapagos, Genentech, GlaxoSmithKline, Human Genome Sciences, Janssen, Eli Lilly, Merck, Mesoblast, Novartis, Novo Nordisk, Pfizer, Roche, UCB Biosciences, Sanofi-Aventis, Takeda and Vertex, and has served on the speaker's bureau for AbbVie, Amgen and UCB. RMS was an employee of Celgene at the time of the study. AV and XZ are current employees of Celgene. PB has received research grants from Celgene.
Ethics approval Institutional review boards of the participating centres.
Provenance and peer review Not commissioned; externally peer reviewed.