Article Text

Abstract
Background Beyond genetics, epigenetics alterations and especially those related to DNA methylation, play key roles in the pathogenesis of autoimmune diseases such as primary Sjögren's syndrome (pSS) and systemic lupus erythematosus. This study aimed to assess the role of methylation deregulation in pSS pathogeny through a genome-wide methylation approach.
Patients and methods 26 female patients with pSS and 22 age-matched controls were included in this study. CD4+ T cells and CD19+ B cells were isolated from peripheral blood mononuclear cells by magnetic microbeads and their genome-wide DNA methylation profiles were analysed using Infinium Human Methylation 450 K BeadChips. Probes with a median DNA methylation difference of at least 7% and p<0.01 between patients and controls were considered significantly differentially methylated.
Results Methylation alterations were mainly present in B cells compared with T cells. In B cells, an enrichment of genes with differentially methylated probes in genetic at-risk loci was observed, suggesting involvement of both genetic and epigenetic abnormalities in the same genes. Methylation alterations in B cells were more frequent in some specific pathways including Interferon Regulated Genes, mainly among patients who were autoantibody positive. Moreover, genes with differentially methylated probes were over-represented in B cells from patients with active disease.
Conclusions This study demonstrated more important deregulation of DNA methylation patterns in B cells compared with T cells, emphasising the importance of B cells in the pathogenesis of the disease. Overlap between genes with differentially methylated probes in B lymphocytes and genetic at-risk loci is a new finding highlighting their importance in pSS.
- Sjøgren's Syndrome
- T Cells
- B cells
- Autoimmune Diseases
- Autoantibodies
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Primary Sjögren's syndrome (pSS), also referred to as autoimmune epithelitis, is a complex systemic autoimmune disease (AID) affecting 0.01% to 0.3% of the general population with a 9/1 female predisposition.1 ,2 Lymphoid infiltration of lacrimal and salivary glands leading to xerophthalmia and xerostomia, as well as enhanced activation of polyclonal B lymphocytes, is the hallmark of the disease. The disease mainly affects the exocrine glands (particularly salivary and lacrimal glands), but one-third of the patients develop several systemic complications, such as renal, pulmonary or neurological manifestations. Around 5% of patients with pSS will develop lymphoma. In spite of the progress in the past 10 years, the pathogenesis of the disease remains to be elucidated.3
Genetic studies represent a powerful tool for the identification of new pathogenic pathways. Results of the first genome-wide association study (GWAS) among Caucasian patients with pSS were recently published.4 The genetic loci most significantly associated with pSS were the major histocompatibility complex/human leucocyte antigen (MHC/HLA) region, IRF5, BLK, STAT4, IL12A, TNIP1 and CXCR5. Therefore, the findings of this first GWAS in pSS highlight the three major pathogenic steps implicated in this disease: activation of the innate immune system, notably through the interferon (IFN) system; B cell activation, through CXCR5-directed recruitment to lymphoid follicles and B cell receptor activation involving Blk; and T cell/NK cell activation owing to HLA susceptibility and the interleukin (IL)-12–IFNγ axis.
Beyond genetics, epigenetics alterations including changes in DNA methylation, histone modifications and microRNA expression probably play key roles in the pathogenesis of AIDs such as pSS.5–7 Although the understanding of epigenetics in pSS is limited at present, distinct salivary gland microRNA expression patterns have been linked to the disease.8 DNA methylation is considered the core epigenetic mechanism that regulates gene expression by altering transcriptional accessibility of gene regulatory regions. Two recent studies investigated DNA methylation changes in naïve CD4+ T cells from patients with lupus and demonstrate that IFN-regulated genes (IRGs) in blood CD4+ T cells from patients with lupus are epigenetically poised for transcription.9 ,10 The first epigenome-wide DNA methylation study in patients with pSS analysing blood naive CD4+ T cells found a similar hypomethylation of a set of IRGs.11
In the present study, we performed a genome-wide DNA methylation study in blood CD4+ T cells and in blood CD19+ B cells and found that DNA methylation changes were significantly more frequent in B cells compared with T cells and were implicating the same genes and pathways found to be associated with the disease in genetic studies. The presence of abnormalities in pathways shared by genetics and epigenetics provides a strong argument for the importance of these pathways in the pathogenesis of the disease.
Patients and methods
Patients
Twenty-six female Caucasian patients with pSS and 22 age- and ethnicity-matched controls recruited in a tertiary national reference centre for pSS in France were included in this study. All patients with pSS fulfilled the American European consensus group (AECG) 2002 criteria for the disease. Activity of the disease was assessed by the recently validated EULAR Sjögren's syndrome Disease Activity Index (ESSDAI) score.12 Controls were either healthy subjects or patients suffering from a mechanical rheumatological condition without any sign of autoimmunity, cancer or inflammation. None were sicca controls. The study was approved by the local ethics committee and written informed consent was obtained from all patients and controls.
Cell isolation and DNA preparation
CD4+ T lymphocytes and CD19+ B lymphocytes were purified from peripheral blood mononuclear cells (PBMCs) of patients and controls by direct magnetic labelling with CD4 and CD19 microbeads (Miltenyi Biotec, Paris, France), and their purity was analysed on a BD FACSCanto (BD Biosciences, San Jose, California, USA) and confirmed to be higher than 95%. DNAs were extracted using the QIAamp DNA Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's protocol.
Genome-wide DNA methylation study and validation of differentially methylated CpG sites by pyrosequencing
DNAs were analysed using the Infinium Human Methylation 450 K BeadChips (Illumina, San Diego, California, USA) according to the manufacturer's protocol. Quantitative DNA methylation analysis for validation was performed by pyrosequencing13 as described in detail in the online supplementary methods.
Statistical analyses
Raw signals of 450 K BeadChips were extracted using the GenomeStudio software (V.2011.1, Illumina) and processed using a refined version of the SQN pipeline14 as described in detail in the online supplementary methods. For the comparison of DNA methylation levels of CpG sites between patients and controls, CpG sites with a methylation difference of more than 7% and a statistical difference of p<0.01 in a non-parametric Mann–Whitney test were considered as statistically significantly differentially methylated. Genes were considered as differentially methylated if two CpG fulfilled these differences. Analyses restricted to the probes located at the gene start and promoter regions were also provided. Potential enrichment of differentially methylated regions in genomic intervals was tested using an in-house developed R script. Significance of the enrichment was estimated based on Monte Carlo simulations.
The most over-represented biological terms and canonical pathways related to the differentially methylated genes were identified using Ingenuity Pathway Analysis (Qiagen) by submitting the gene lists as defined above. A Benjamini–Hochberg corrected p value <0.05 was considered significant. The Interferome database15 (http://interferome.its.monash.edu.au/interferome/home.jspx) was used for the analysis of the possible involvement of significantly differentially methylated IRGs in pSS. In addition, a manually curated list including ∼100 IRGs based on the published literature was used for the same purpose.10 ,16–22
Results
Twenty-six female Caucasian patients with pSS and 22 age- and ethnicity-matched controls were included in this study (see online supplementary table S1). Seventy-three per cent of the patients had anti-SSA antibodies and 42% anti-SSB antibodies. All patients producing anti-SSB antibodies were also anti-SSA positive. Mean ESSDAI was 3.5±4. The genome-wide DNA methylation analysis using the 450 K BeadChips showed large differences in DNA methylation patterns between CD19+ B lymphocytes and CD4+ T lymphocytes from patients with pSS with more than 60 000 CpG sites being differentially methylated.
Methylation status of purified blood T cells from patients with pSS
Among purified CD4+ T cells, only 119 probes were detected in patients compared with the control cohort to be differentially methylated at a threshold of 7% (see online supplementary data file). These probes were distributed across 74 genes; 40 genes (54%) were hypermethylated, 33 genes (45%) were hypomethylated and a single gene displayed a mixed methylation pattern (figure 1A). Forty-three probes were found when the DNA methylation analysis was restricted to gene start and promoter regions (where DNA methylation changes might correlate inversely with the gene expression potential). These probes were distributed across 37 genes including 18 hypermethylated genes (49%) and 19 hypomethylated genes (51%) (figure 1B). Furthermore, seven genes were differentially methylated at two or more CpGs (figure 1C), four of which in the promoter regions (figure 1D).

Methylation status of purified lymphocyte T cells from patients with primary Sjögren's syndrome (pSS) compared with controls. (A) Probes with at least 7% median difference of methylation and p<0.01 found in T lymphocytes from patients with pSS compared with the control cohort and number of genes with at least one probe differentially methylated. (B) Probes in the TSS/promoter region with more than 7% median difference of methylation and p<0.01 found in T lymphocytes from patients with pSS compared with the control cohort and number of genes with at least one probe differentially methylated. (C) Number of the genes with a minimum of two probes using the same thresholds. (D) Number of genes with minimum two probes in TSS/promoter regions with more than 7% median difference and p<0.01. Genes with hypermethylated probes are denoted with ‘+’; genes with hypomethylated probes with ‘−’; genes with hyper and hypomethylated probes are annotated as mixed genes. Similar representations for other methylation thresholds are shown in online supplementary figure S1.
Methylation status of purified blood B cells from patients with pSS
In B lymphocytes, out of a total 6707 probes, 3754 hypermethylated (56%) and 2953 (44%) hypomethylated probes were found in patients compared with the control cohort. These probes were distributed across 3619 genes including 1972 (55%) genes with only hypermethylated CG sites, 1389 (38%) genes with only hypomethylated CG sites and 258 (7%) genes with mixed methylation pattern of CpG sites (figure 2A). Among them, 2393 probes were located in the gene start/promoter regions (figure 2B). Eight hundred and ninety-nine genes were differentially methylated at two or more CpG positions: 363 hypermethylated (40%), 278 hypomethylated (31%) and 258 (29%) displaying mixed methylation pattern (figure 2C). A total number of 391 genes showed differential methylation in the gene start/promoter regions corresponding to 162 hypermethylated (42%), 173 hypomethylated (44%) and 56 genes (14%) displaying a mixed methylation pattern (figure 2D). Increasing the methylation difference to 10%, 15% or 20% did not change the proportions significantly (see online supplementary figure S1). The differentially methylated genes detected with different methylation thresholds are presented in the online supplementary data file. Consistent with the similar numbers of hyper and hypomethylated probes/genes found in the genome-wide study, no change of the global methylation status was observed by pyrosequencing analysis of multicopy repetitive elements (ALU and LINE1) (data not shown). Subsequently, the location of the differentially methylated CpG sites in relation to genomic elements such as CpG islands (islands, shores, shelves and other) or gene structure (TSS1500, TSS200, 5′UTR, first exon, gene body, 3′UTR and intergenic) was analysed to investigate a differential representation of functional categories (see online supplementary figure S2). The differentially methylated probes in CD19+ cells from patients with pSS had a similar repartition in shores and shelves when compared with the overall proportion of these probes on the BeadChip, but were significantly depleted in CpG islands (14.6% vs 30. 9% of total probes; p<0.05 Monte Carlo simulation, n=5000) and enriched outside CpG islands (47.74% vs 36.38% of total probes, p<0.05). No significant difference was found with the location of differentially methylated CpG sites in relation to gene structure (see online supplementary figure S2). To investigate if DNA methylation alterations could cluster at specific chromosomal loci such as the MHC, an enrichment analysis was performed using the positions and density of probes on the array as background. However, no loci were identified as enriched (data not shown).

Methylation status of purified B lymphocytes from patients with primary Sjögren's syndrome (pSS) compared with controls. (A) Probes with at least 7% median difference of methylation and p<0.01 found in B lymphocytes from patients with pSS compared with the control cohort and number of genes with at least one probe differentially methylated. (B) Probes in the TSS/promoter region with more than 7% median difference of methylation and p<0.01 found in B lymphocytes from patients with pSS compared with the control cohort and number of genes with at least one probe differentially methylated. (C) Number of the genes with a minimum of two probes using the same thresholds. (D) Number of genes with minimum two probes in TSS/promoter regions with more than 7% median difference and p<0.01.
Validation of CpG methylation changes by pyrosequencing
To validate the results from the 450 K BeadChip, we quantitatively analysed DNA methylation patterns of the CpGs of interest in B lymphocytes by pyrosequencing (figure 3A). All DNA methylation alterations in IFN-related genes including IFITM1, IFITM3, IFI44 L, IRF5 were validated by pyrosequencing with a p value<0.05 (figure 3B). Other genes of potential interest for their implication in the disease pathogenesis (RUNX3, TNFAIP8, IKZF1, SLC15A4, GRB2, MIR21, IL21R, CXCR5, and TRAF5) found with significantly differentially methylated probes were also confirmed by pyrosequencing in patients with pSS compared with controls (figure 3A) demonstrating the confidence in the obtained gene lists.
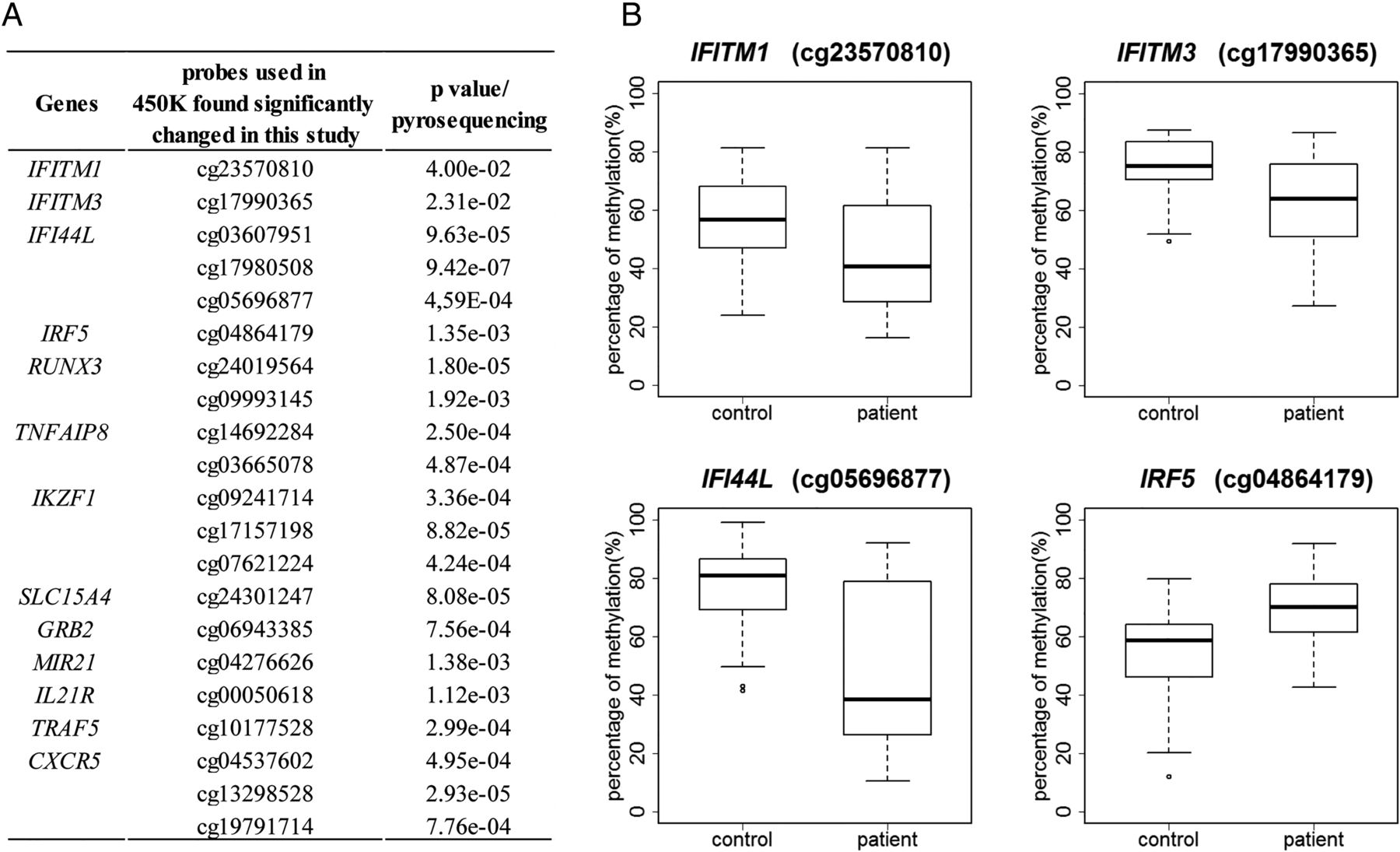
Validation of DNA methylation changes in different genes by pyrosequencing. (A) A selection of genes found significantly differentially methylated in 450 K BeadChip and validated by pyrosequencing. (B) Boxplots of DNA methylation levels of several representative interferon regulated genes by pyrosequencing are shown.
Pathway analysis
To identify pathways possibly influenced by the differential methylation in B lymphocytes from patients with pSS compared with controls, we performed a pathway analysis using only genes with a minimum of two differentially methylated CpG sites. IL-4, IL-8, CXCR4, PTEN and B cell receptor signalling, B cell development, altered T cell and B cell signalling in rheumatoid arthritis and systemic lupus erythematosus (SLE) signalling were identified as enriched pathways (figure 4). Differentially methylated genes were enriched for chronic inflammatory disease (p=6.76×10−7), arthritis (p=3.16×10−6), rheumatic disease (p=1.31×10−5), systemic autoimmune disorder (p=1.24×10−4), lymphoid cancer (p=1.26×10−4), lymphomagenesis (p=1.46×10−4) as disease annotation. Furthermore, as ∼5% of patients with pSS develop lymphoma, gene lists were investigated for genes associated with B-cell lymphoma and 22 significantly differentially methylated genes involved in lymphoma including TNFRSF10A, IRF8, CXCR5, LTA, SOCS1, TNF, BCL2, PSMB2, PIK3CD (p=5.37×10−4) were found.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pathway analysis of the differentially methylated genes in CD19+ B cells from patients with pSS or from sub-groups of patients. The dashed line represents a p value of 0.05. BL, Total B lymphocytes; IL, interleukin; pSS, primary Sjögren's syndrome; RA, rheumatoid arthritis; SLE, systemic lupus erythematosus.
Furthermore, although not differentially methylated themselves, several proteins and cytokines, including some which have been previously implicated in AIDs, were identified as upstream regulators, whose deregulation could be at least partially responsible for the observed effects at the DNA methylation level: IL27 (p=3.54×10−5), IFNγ (p=0.002), IFNα (p=0.002), CD40 L (p=0.001), TLR3 (p=9.11×10−4), TLR7 (p=0.006) as well as the oestrogen receptor (p=7.71×10−5).
Overlap between genome-wide DNA methylation analysis and genetic at-risk loci
A GWAS performed in American and European patients with pSS showed that the HLA region on chromosome 6 was strongly associated with pSS.4 Furthermore, single nucleotide polymorphisms (SNPs) in or close to IRF5-TNPO3, STAT4, IL12A, FAM167A-BLK, DDX6-CXCR5 and TNIP1 were also strongly associated with pSS; while TNFAIP3, DGKQ, ITSN2 showed associations with suggestive significance. Investigating the overlap between the genes having at least one differentially methylated probe and the GWAS at-risk loci, 7/17 GWAS as at-risk loci (HLA-DRA, HLA-DQB1, IFR5, CXCR5, BLK, PRDM1, ITSN2) for patients with pSS showed at least one significantly differentially methylated probe (table 1A). Similarly, the overlap was investigated with respect to the GWAS performed in Han Chinese patients with pSS, and of the seven genes identified in this study, four (GTF2I, HLA-DQA1, HLA-DPB1, COL11A2) were also found with at least one significantly differentially methylated probe in B cells from patients with pSS (table 1B).23
Genes with significantly differentially methylated probes found at GWAS at-risk loci
Association of DNA methylation with disease activity in B lymphocytes from patients with pSS
To correlate DNA methylation with disease activity, we analysed the methylation status of B lymphocytes from patients with pSS with high (≥5) or low (<5) ESSDAI score compared with the control cohort. Among the patients with pSS with high ESSDAI score, 5687 genes were detected with at least one significantly differentially methylated probe, while among those with low ESSDAI score, much fewer genes (n=1772) were found (see online supplementary figure S3). Some of the above described canonical pathways like IL-8 and CXCR4 signalling (figure 4) were only significantly enriched in patients with high ESSDAI scores , while B cell development and altered T cell and B cell signalling in rheumatoid arthritis was only found in patients with low ESSDAI scores.
Patients with pSS were also divided into three sub-groups based on the production of anti-SSA and anti-SSB autoantibodies. In B lymphocytes from patients who were anti-SSA negative, only 535 genes were found with at least one significantly differentially methylated probe (see online supplementary figure S4), while B lymphocytes from patients who were anti-SSA+/anti-SSB− and anti-SSA+/anti-SSB+ pSS showed 1798 and 3674 genes, respectively. The identified significantly enriched pathways shown in figure 4 were only found in autoantibody producing subgroups (figure 4).
As IFNα and IFNγ were found as predicted upstream regulators of the observed changes in this study, patients with pSS were reported to show an IFN signature16 and IPA has been reported to have some shortcomings in the identification of an IFN signature,10 we submitted the gene lists with at least two differentially methylated CpG positions to the interferome database15 to identify IRGs. Compared with the controls, B lymphocytes from patients with pSS producing only anti-SSA antibodies, or both anti-SSA and anti-SSB, showed a similar proportion of differentially methylated IRGs (9.9% vs 10.9%), whereas this proportion dropped significantly (4.3%) in B lymphocytes from patients with pSS without autoantibodies (see table 2 and online supplementary table S2). The difference in IFN signatures between the patients producing autoantibodies and the seronegative patients was also confirmed by crossing our gene lists with a manually curated IRG list (data not shown). All these results lead to the conclusion that in B lymphocytes the level of methylation of a large number of genes including those involved in the IFN signature is associated with disease activity in patients with pSS.
Number and percentage of significantly differentially methylated IFN-regulated genes found in B lymphocytes from patients with pSS who were autoantibody positive or negative compared with the control cohort, respectively
Discussion
This genome-wide methylation study on cell sorted PBMC populations of patients with pSS revealed that methylation alterations are mainly present in B cells compared with T cells. In contrast to previous reports on DNA methylation changes in AIDs,24 we did not observe a global hypomethylation in our study with relatively equal number of hypermethylated and hypomethylated genes as well as an absence of changes of DNA methylation patterns at repetitive elements.
Very interestingly, 40% of genes genetically associated to the disease were detected with at least one differentially methylated CpG site, suggesting the involvement of both genetic and epigenetic abnormalities in the same genes and emphasising the role of these genes in the pathogenesis of the disease. The mechanisms causing AIDs are poorly understood. Different cells might play a biased role in the development of different AIDs. Our study is the first to analyse genome-wide DNA methylation patterns in B cells of patients with pSS . Recently, Altorok et al reported the methylation deregulation in naïve CD4+ T cells from patients with pSS using the same technology as in our study.11 They identified about 426 differentially methylated genes with three quarters of them being hypomethylated. This number is much larger than the genes identified in the current study (n=74). Of note, in T cells, four hypomethylated (STAT1, SH3PDX2A, FLI37453, USP18) and one hypermethylated gene (FBXL16) were found in both studies. The observed difference might be due to a difference in the analysed cell population as Altorok et al analysed only naïve CD4+ T cells, while ours analysed all CD4+ cells as well as differences in the patient population such as a younger age in the Altorok study and different prevalence of autoantibodies, different treatments and different disease activity, which might influence T cell populations. Furthermore, the time point in the disease course, at which sampling has occurred, or lifestyle factors might have an impact on the DNA methylation profile.
Analysis of genome-wide DNA methylation patterns in CD4+ T cells, CD19+ B cells and CD14+ monocytes from patients with SLE10 showed six times more DNA methylation changes in T cells compared with B cells and ten times more than in monocytes, in concordance with the hypothesis that T lymphocytes play a central role in the development of SLE.25 Our results show that the situation is different in pSS with more DNA methylation alterations present in B lymphocytes compared with T cells. These findings are in concordance with several recent reports showing that B cells may play a central role in the development of pSS.3 ,26 Only few markers differentially methylated in both T cells and B cells were identified in our study, underlining the importance that methylation studies have to be conducted in well-characterised pure cell populations27 and that the choice of the selected cell population is of critical importance to decipher a disease. It would have been desirable to analyse even more specific subpopulation of the B cells, which might show a more pronounced epigenetic deregulation such as naïve B cells, memory B cells or plasmablasts. However, the quantity of blood required for the 450 K BeadChips cannot be reasonably obtained from patients and controls. The observed DNA methylation changes should however not be due to a shift in the subpopulations as the total number of lymphocytes is decreased in patients with pSS, but the percentage of B cells, naïve B cells, memory B cells, CD4, CD8 and NK cells is the same as in controls (Mariette, unpublished data).
Differential DNA methylation in B lymphocytes was found among other loci in HLA-DRA, HLA-DQB1, IRF5, CXCR5, BLK, PRDM1, ITSN2 which were all identified in a recent GWAS as at-risk loci,4 suggesting that both genetic and epigenetic alterations in the same genes closely interact in the development and course of AIDs emphasising the probable important role of these genes in the pathogenesis of the disease. Similar observations have been previously reported in other AIDs such as rheumatoid arthritis with multiple interactions between genetic and epigenetic variants in the MHC region28 or lupus.29
We previously investigated in a gene-specific study the promoter region of IRF5 since an SNP associated with pSS consisted of a four times repeat of a CGGGG motif which is repeated only three times in the wild type allele.30 As this polymorphism introduces a supplementary CpG, we investigated CpGs around this location as well as the transcription start site of IRF5, but found no significant differences. In the present study, we confirm the lack of a significant difference between patients with pSS and controls regarding the previously studied CpGs, but identified differentially methylated CpG in IRF5 locus 400 bp apart from the previously studied CpGs and closer to the locus of genetic association, which is ∼2 kb further upstream. This observation reinforces the usefulness of tools that assess the methylation with a large number of probes like the 450 K BeadChip.
The genes with significantly different DNA methylation patterns in our study were enriched for several pathways related to B cell signalling, inflammation and AIDs, further emphasising the involvement of disease-relevant changes in B cells. Significant changes in the DNA methylation patterns observed in CD19+ cells from patients with pSS included a number of IRGs, which were also confirmed by pyrosequencing of IFITM1, IFITM3, IFI44L, IRF5, TNFAIP8, IKZF1. Furthermore, we found more genes with differentially methylated CpG sites in B cells from patients with active disease as defined by the ESSDAI score and in patients who were auto-antibody positive and the DNA methylation signature of IRGs was more prominent in the patients who were autoantibody positive. Microarray-based gene expression studies on labial salivary glands and peripheral blood showed that in patients with pSS type I IFN-inducible genes were deregulated.16 ,31 Cross-talk of the IFN type I pathway with the IFN type II pathway reciprocally has been found to affect each other's production and signalling in patients with AID.32 ,33 The significant changes in the DNA methylation patterns observed in CD19+ cells from patients with pSS also included a number of IRGs, which also correlated with the autoantibody status. Similar findings have previously been made in type 1 diabetes, where disease-specific markers were also present in autoantibody positive individuals that were not yet showing overt clinical symptoms of the disease.34
In summary, we performed a comprehensive genome-wide DNA methylation analysis in patients with pSS in blood-derived T and B cells and demonstrated more important DNA methylation deregulation in B cells than in T cells, emphasising the importance of B cells in the pathogenesis of the disease. Altered DNA methylation patterns were found in the same genes previously reported to contain genetic variation associated with the disease, which suggests the importance of combined analysis of genetic and epigenetic variations to identify new targets involved in the pathogenesis of AIDs. While this study as well as the previously published study by Altorok et al11 provide new avenues for research in pSS and AIDs in general, larger studies as well as the analysis of additional cell types are required to gain a more comprehensive understanding of DNA methylation changes implicated in the pathogenesis. In addition, other epigenetic mechanisms influencing gene expression, such as abnormal expression of microRNAs, which has been found to be involved in pSS in specific cellular populations,8 will also help identifying new therapeutic targets in this disease.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online data
- Data supplement 3 - Online figures
- Data supplement 4 - Online tables
Footnotes
Handling editor Tore K Kvien
CM-R and S-FW-R contributed equally. XM and JT are two senior authors and contributed equally.
Contributors CM-R, XM and JT designed the study; CM-R, SB, RB, GN and XM collected patient material and assessed clinical parameters; SB, FB, CL and KB performed molecular experiments; CM-R, S-FW-R, FB, XM and JT analysed the data; CM-R, S-FW-R, XM and JT drafted the manuscript. All authors read and accepted the final version of the manuscript. CM-R and S-FW-R contributed equally to the study. XM and JT contributed equally to the study.
Funding The research described in this manuscript was funded by a grant from the French National Research Agency (ANR- 2010-BLAN-1133 01).
Competing interests None declared.
Ethics approval CCP Ile de France VII No. CO-10-003 on 3rd February 2010.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Normalised or raw data of the 450 K BeadChips is available upon request from the authors.