Article Text

Abstract
Objective An interesting and so far unexplained feature of chronic pain in autoimmune disease is the frequent disconnect between pain and inflammation. This is illustrated well in rheumatoid arthritis (RA) where pain in joints (arthralgia) may precede joint inflammation and persist even after successful anti-inflammatory treatment. In the present study, we have addressed the possibility that autoantibodies against citrullinated proteins (ACPA), present in RA, may be directly responsible for the induction of pain, independent of inflammation.
Methods Antibodies purified from human patients with RA, healthy donors and murinised monoclonal ACPA were injected into mice. Pain-like behaviour was monitored for up to 28 days, and tissues were analysed for signs of pathology. Mouse osteoclasts were cultured and stimulated with antibodies, and supernatants analysed for release of factors. Mice were treated with CXCR1/2 (interleukin (IL) 8 receptor) antagonist reparixin.
Results Mice injected with either human or murinised ACPA developed long-lasting pronounced pain-like behaviour in the absence of inflammation, while non-ACPA IgG from patients with RA or control monoclonal IgG were without pronociceptive effect. This effect was coupled to ACPA-mediated activation of osteoclasts and release of the nociceptive chemokine CXCL1 (analogue to human IL-8). ACPA-induced pain-like behaviour was reversed with reparixin.
Conclusions The data suggest that CXCL1/IL-8, released from osteoclasts in an autoantibody-dependent manner, produces pain by activating sensory neurons. The identification of this new pain pathway may open new avenues for pain treatment in RA and also in other painful diseases associated with autoantibody production and/or osteoclast activation.
- Ant-CCP
- Autoantibodies
- Rheumatoid Arthritis
- Fibromyalgis/Pain Syndromes
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a common chronic autoimmune disease that clinically is typified by swelling and reduced function in affected joints, and with joint pain as one of the dominant symptoms.1 A so far unexplained feature of chronic pain in RA is the frequent disconnect between pain and inflammation. Joint pain (arthralgia) often develops before signs of joint inflammation and is thus one of the first indicators of an emerging RA. Interestingly, autoantibodies also frequently occur in the preclinical phase of the disease and can be detected months to years prior to diagnosis.2–4 Further, recent studies show that despite reduced disease activity in response to treatment with disease-modifying antirheumatic drugs, many patients continue to report mild, moderate or severe pain.5–7 However, the mechanisms that are responsible for the inflammation-independent pain that occurs both in the very early phase of the disease and in periods of medically controlled disease activity8 are so far unknown. Here, we address the possibility that certain autoantibodies directed towards post-translational (citrullinated) proteins may drive the non-inflammatory pain associated with RA.
Anticitrullinated protein antibodies (ACPA) are present in a major subset of patients with RA and are used clinically as serological markers for the diagnosis of RA.9 Citrullinated autoantigens in patients with RA include fibrinogen, vimentin, α-enolase, collagen type II, immunoglobulin-binding protein and histone 4.10 Although ACPA is associated with arthralgia before the onset of inflammation and a more aggressive RA subsequent to clinical diagnosis, relatively little is known about the potential pathogenic effects of the human ACPA response.11 It has been suggested that ACPA together with other pathogenic antibodies facilitate the development of experimental arthritis in mice.12 ,13 Furthermore, recent evidence show that certain ACPA alone can induce alterations in bone metabolism and bone loss after binding to the cell surface of osteoclasts.14
Previous studies on the contribution of autoantibodies to pain have predominantly been concerned with their role in inflammatory processes, leading to activation of inflammatory cells and the subsequent release of pain-inducing cytokines and prostaglandins. Recent work, however, shows that autoantibodies against neuronal voltage-gated potassium channels induce pain without signs of inflammation,15 suggesting that autoantibodies may in some contexts have a role in chronic pain states also in the absence of apparent inflammation. The aim of the present study was to examine whether ACPA can induce and maintain joint pain, and whether this is coupled to joint pathology.
Materials and methods
Detailed information on materials and methods is available as online supplementary file.
Animals and injections
Experiments were performed using male B10.RIII mice (Department of Medical Biochemistry and Biophysics, Karolinska Institutet) and BALB/c (Harlan) 15–22 weeks of age. Mice were housed in standard cages (3–5 per cage) in a climate-controlled environment maintaining a 12 h light/dark cycle with access to food and water ad libitum. All experiments were approved by the local ethics committee for animal experiments in Sweden (Stockholm Norra Djurförsöksetiska nämnd). Mice were injected intravenously with either saline or human IgG (hACPA and controls 0.125–4 mg), monoclonal murinised ACPA antibodies (mAb; 2 mg, single Ab or D10 and B2 1:1). Intra-articular injection was performed under isoflurane anaesthesia. A 1:1 mix of 30 ng CXCL1 and CXCL2 (Sigma) or 30 ng of each was injected into the ankle joint. The CXCR1/2 antagonist reparixin (MedChem Express) was injected subcutaneously twice daily (30 mg/kg/day).
Preparation of human ACPA (hACPA, anti-CCP2 IgG antibodies)
The patients were diagnosed according to the 1987 ACR criteria16 and determined to be ACPA positive (ACPA+) or ACPA negative (ACPA−) using a routine assay for ACPAs (CCP2 assay). Purification of IgG from plasma and sera from patients with ACPA+ RA, ACPA− RA and healthy age-matched donors was done as described previously using HiTrap Protein G columns (GE Healthcare).17 ACPA+ IgG was then further purified using CCP2 affinity column (Euro-Diagnostica). IgG not binding to the CPP2 column was used as control denoted by flow through (FT).
Generation of monoclonal ACPA
Murinised monoclonal D10, B2, C7 and E2 (control) IgG2a were generated as previously described.18 B cells from the synovial fluid of patients with RA were single sorted, cloned into expression vectors and selected for reactivity for citrullinated epitopes (CEP-1, vimentin and fibrinogen) or control epitope (human tetanus).
Mechanical and thermal hypersensitivity
Withdrawal thresholds of the hind paws were assessed using von Frey filaments as previously described.19 A 50% withdrawal threshold was calculated using the Dixon up–down method,20 and the results are presented as per cent of baseline values.
Heat sensitivity was examined using a modified Hargreaves box21 and cold sensitivity by gently applying a drop of acetone to the hind paw and measuring the duration of nocifensive behaviour (lifting, shaking, biting and licking the paw). The tests were repeated three times on each paw and the average calculated.
Locomotor activity and food/water consumption
Activity level of the mice during a full night cycle was measured using Oxymax/Comprehensive Lab Monitoring System (Columbus Instruments). Mice were monitored during the night cycle (18:00–06:00). Infrared sensors detect movement in X, Y and Z axes and the number of beam breaks recorded and presented as total movement (number of XY-axis beam breaks), ambulation (number of consecutive XY-axis beam breaks) and rearing (number of beam breaks in the Z axis).
Tissue analysis
Metalloprotease activity, western blot, histology, immunohistochemistry, qPCR on tissues and dorsal root ganglion (DRG) functional studies are described in online supplementary methods.
Osteoclast cultures and chemokine analysis
For in vitro osteoclasts generation, bone marrow cells were obtained from wildtype BALB/c mice (Harlan), and CD-11b+ cells were cultured with M-CSF and RANKL (both Peprotech). Levels of CXCL1 (KC-GRO, Meso Scale Discovery) and CXCL2 (MIP-2α, R&D systems) were measured in the supernatants.
Statistical analysis
For comparing changes over time, repeated measures two-way analysis of variance (ANOVA) was used followed by Bonferroni post hoc test. For differences in three groups or more, one-way ANOVA was used, followed by Bonferroni post hoc test. For differences in two groups, Student’s t test was used. Arthritis and histological scores were compared using the Kruskal–Wallis test followed by Dunn's multiple comparison post hoc test. All tests were performed using GraphPad Prism 6 software. p Values <0.05 were considered significant.
Results
Antibodies from human patients with RA induce pain-like behaviour in mice
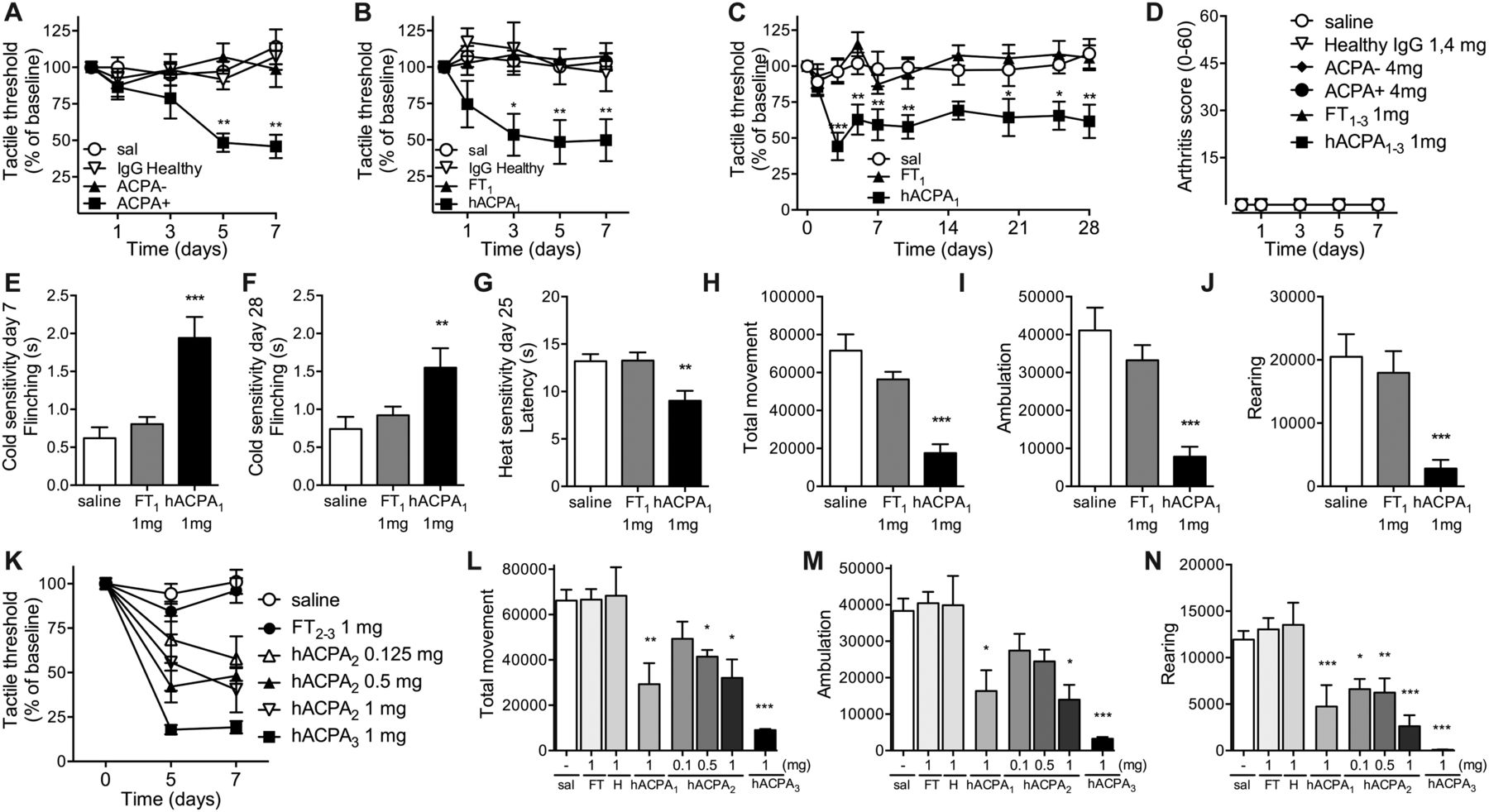
To examine if RA-associated autoantibodies are directly linked to nociception (pain), we assessed pain-like behaviour in mice after intravenous administration of IgG isolated and pooled from patients with RA or from healthy age-matched donors. A pronounced drop in tactile thresholds was observed in mice injected with 4 mg IgG from patients with ACPA+ RA, but not from patients with ACPA− RA or healthy individuals (figure 1A).

Mechanical and thermal sensitivity and locomotor activity in mice following injection of human antibodies. Mechanical sensitivity in mice injected intravenously with saline (sal), IgG from healthy donors, IgG from patients with ACPA− RA or IgG from patients with ACPA+ RA (4 mg, n=9/group) (A) and purified human (h) ACPA IgG (batch 1, 1 mg, n=4), non-ACPA IgG from the same patients (FT, 1 mg, n=6) and IgG from healthy donors (1 mg, n=6) (B). ACPA and FT from batch 1 were injected into a different strain of mice (n=7/group) and mechanical sensitivity assessed over time (C), cold sensitivity days 7 and 28 (E and F) and heat sensitivity day 25 (G). Total movement (H), ambulatory (directional) movement (I) and rearing (J) were monitored 12 h the third night (same mice as in C). Arthritis scores (0–60) (D). Mechanical sensitivity days 5 and 7 (K) and total movement (L), ambulatory movement (M) and rearing (N) during third night after injection with 1 mg ACPA batch 1–3 (n=3 each) or 0.5 mg (n=7), 0.125 mg (n=6) ACPA batch 2 or corresponding FT (n=6/group) or saline. Data are presented as mean±SEM. *p<0.05, **p<0.01, and ***p<0.001 are compared with saline. ACPA, anti-citrullinated protein antibodies; FT, flow through; RA, rheumatoid arthritis.
Pain-like behaviour in mice is only induced by the ACPA fraction of IgG
Three batches (batch 1–3) of affinity-purified ACPA and corresponding FT fractions were prepared, containing ACPA IgG from 38, 6 and 25 patients with ACPA+ RA, respectively. Strikingly, while FT IgG (batch 1, 1 mg) and IgG from healthy individuals (1 mg) injected intravenously did not alter the thresholds for evoking a response to mechanical stimulation, ACPA (batch 1, 1 mg) induced mechanical hypersensitivity within 3 days (figure 1B, C), which lasted for at least 28 days (figure 1C). The ACPA-injected mice displayed increased heat and cold sensitivity compared with saline-injected and FT-injected mice (figure 1E–G). Spontaneous (non-evoked) pain-like behaviour was also assessed. Total movement, ambulation (directional walking) and rearing were monitored in the night between days 2 and 3. Similar assays have been used successfully to study non-reflexive pain-like behaviour in experimental models of pain.22 Injection of 1 mg purified ACPA (batch 1), but not 1 mg FT IgG (batch 1), induced a reduction in all movement parameters (figure 1H–J). No signs of joint swelling (figure 1D) or inflammation-related sickness behaviour (piloerection, weight loss, reduced feeding, online supplementary figure S1A–C) were observed, thus the reduction in movement is unlikely to be the result of a local or generalised inflammatory event, but rather the consequence of pronociceptive actions of ACPA. Similar results, with a dose-dependent pain-like behaviour, were obtained with the two other batches of affinity-purified ACPA, in two different mouse strains, and also here no effects were seen with FT IgGs (see figure 1K–N and online supplementary figure S1D). These data indicate that the pronociceptive effect of ACPA is neither restricted to a particular batch of ACPAs nor specific for a particular mouse strain.
Monoclonal mouse ACPA induce pain-like behaviour in mice
D10, B2 and C7 murinised IgG2a cloned from single synovial B cells from human patients with RA18 with varying reactivities for major citrullinated epitopes in RA, but unreactive with the corresponding arginine-containing peptide, were used. Without generating any signs of inflammation (figure 2B), both the murinised D10 and B2 ACPA increased mechanical sensitivity (figure 2C, D) while the C7 ACPA (figure 2C) and E2 (control antibody, figure 2D) did not.

Mechanical sensitivity following injection of murinised monoclonal ACPA in mice. Specificities of the monoclonal antibodies derived from B cells of human patients with RA measured with ELISA,18 using CEP-1, fib36–52, vim60–75 and CCP peptides. Control antibody E2 binds human tetanus (A). Visual inflammation score (0–60) for all monoclonal antibodies (B). Two milligrams of D10 (n=12) (C), C7 (n=7) (C), B2 (n=7) (D), control antibody E2 (n=7) (D) or saline (sal, n=18) were injected and mechanical sensitivity was measured over 20 days. Data are presented as mean±SEM. *p<0.05; **p<0.01 and ***p<0.001 are compared with saline. ACPA, anti-citrullinated protein antibodies; RA, rheumatoid arthritis.
ACPA accumulate in skin, ankle joint and bone marrow
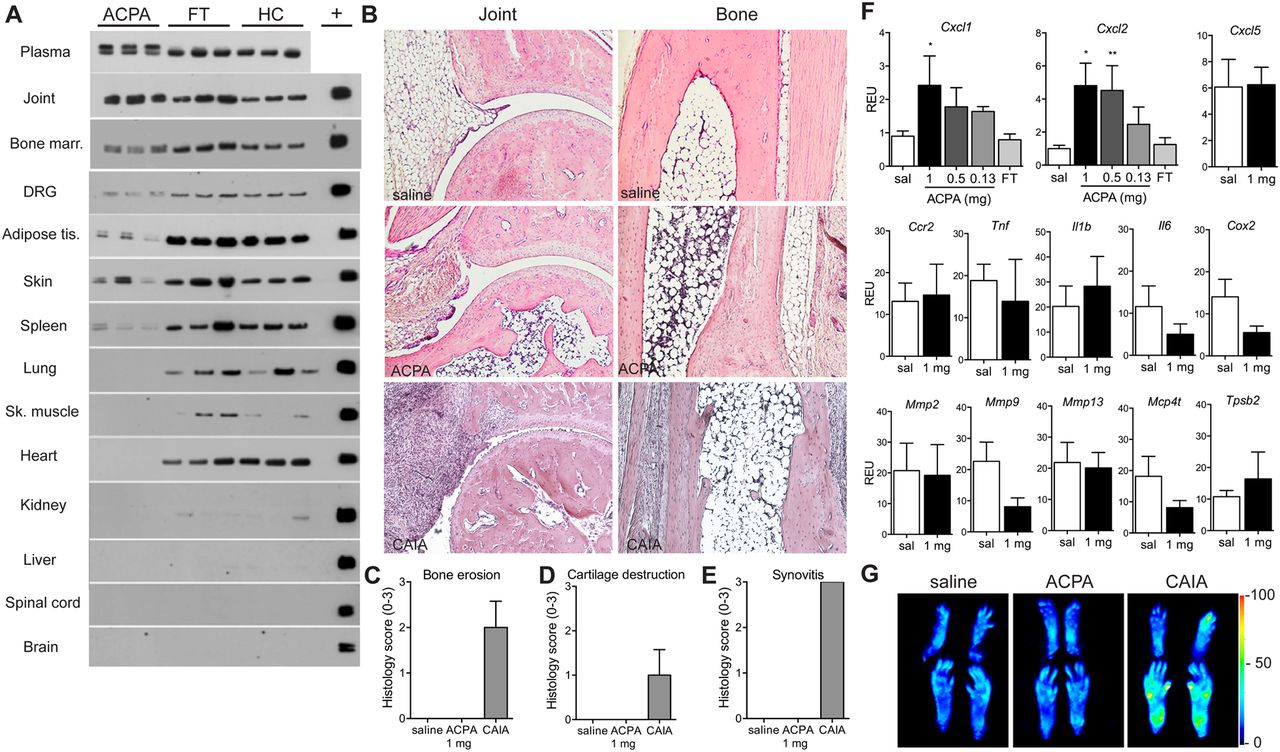
Human ACPA were readily detected in skin, ankle joint, tibial bone marrow and plasma and to some extent in DRG, adipose tissue and spleen, but not in brain or spinal cord (figure 3A) 7 days after injection. Antibodies from healthy controls and FT showed a similar but wider distribution (figure 3A).

Location of antibodies, histology and gene expression in mice after injection of human ACPA. Mice were perfused with saline (sal) to remove blood, and the presence of human IgG in different tissues 7 days after intravenous injection of 1 mg of ACPA3, FT3 or IgG from healthy control (HC) was assessed by western blot. Plasma was used as the positive control (A). Representative ankle joint and tibial bone sections stained with H&E 7 days after injection of human ACPA3 (n=3), saline (n=4) or 15 days after induction of collagen antibody-induced arthritis (CAIA, positive control) (n=3) (B) were scored for bone erosion (C), loss of cartilage (D) and synovitis (E). Ankle joint extracts were analysed by qPCR for changes in mRNA levels 7 days after injection of human ACPA2–3 (n=6) or saline (n=6) and data expressed as relative expression unit (F). Fluorescence image of paws (G), presented as a heat map after intravenous injection with MMPsense680, which becomes fluorescent in the presence of active MMPs in mice injected with saline, human ACPA3 or anticollagen antibodies as the positive control (n=3/group). Data are presented as mean±SEM; *p<0.05 and **p<0.01 are compared with saline. ACPA, anti-citrullinated protein antibodies; FT, flow through.
ACPA does not induce signs of joint inflammation
Examination of histological sections from ankle joints and tibia 7 days after injection of ACPA, FT and saline, did not display any signs of cell infiltration or synovial hyperplasia (figure 3B–E). No difference in mRNA levels between ACPA-injected and saline-injected mice was observed for the chemokines (Cxcl5 and Ccl2), cytokines (Tnf, Il1b and Il6), inflammatory enzyme (Cox2), matrix metalloproteases (Mmp 2, 9, 13) and mast cell proteases (Mcpt4 and Tpsb2; figure 3F). Noteworthy, however, Cxcl1 and Cxcl2 mRNA levels were elevated in ankle joints from ACPA, but not in FT or saline-injected mice (figure 3F). None of the examined factors were elevated in the skin (see online supplementary figure S2A). ACPA did not induce activation of MMPs in the paws (see figure 3G and online supplementary figure S2B).
ACPA does not increase neuronal excitability in neuronal DRG cultures
To investigate if ACPA have a direct effect on peripheral sensory neurons, we investigated the effects of ACPA on Ca2+ fluxes in primary cultures of DRG neurons. Stimulation with FT and ACPA (both 1 µg/mL), followed by KCl (50 mM) to detect cells that can depolarise (ie, neurons) showed an increased intracellular Ca2+ signal in 188 cells in response to KCl. Of the KCl responding cells, ACPA and FT stimulation activated six and four cells (2.5% and 1.7%), respectively (figure 4A, B). Thus, the application of ACPA as well as FT had minor effects on Ca2+ fluxes, and no difference in response between ACPA and FT was detected.
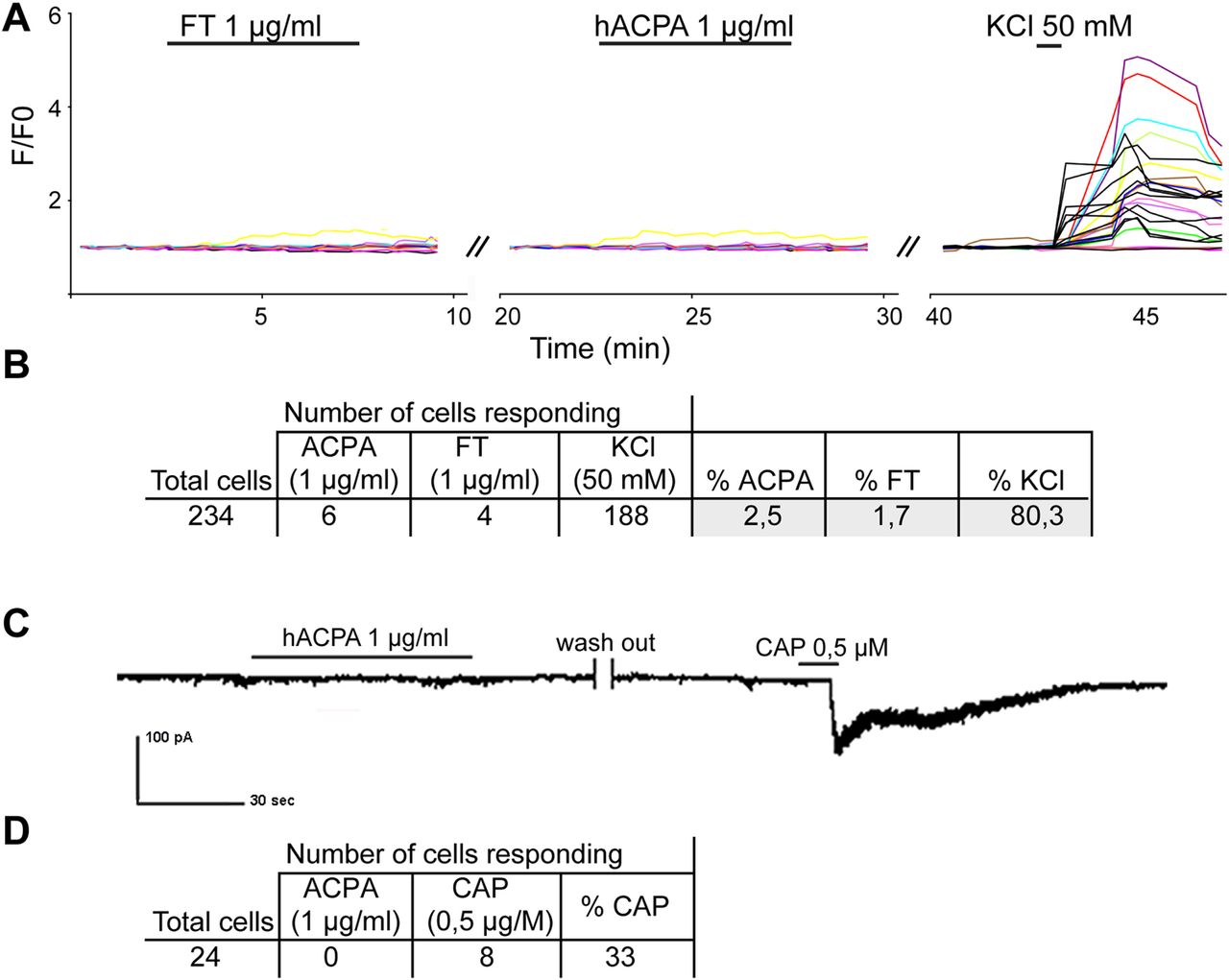
Effect of ACPA on primary peripheral neurons. Mouse dorsal root ganglions were cultured and stimulated with ACPA or FT (both 1 µg/mL). A representative trace showing Ca2+ during stimulation with antibodies and KCl (50 mM) (A). Calcium signal were recorded from 243 cells, where few cells showed a minor response to stimulation (2.5% for ACPA and 1.7% for FT) (B). A total of 24 cells were patched and ionic currents were recorded in whole-cell voltage clamp mode (C). None (0/24) of the recorded cells gave inward current response to ACPA, while 33% (8/24) gave response to capsaicin (1 μM) (D). ACPA, anti-citrullinated protein antibodies; FT, flow through.
Electrophysiological recordings in a subpopulation of small diameter nociceptive neurons that express TRPV1 receptors were undertaken using the TRPV1 agonist capsaicin (0.5 µM) at the end of each experiment for verification. A total of 24 cells were patched and recorded in whole-cell voltage clamp mode. Of the 24 cells, 8 cells gave inward current response to capsaicin (33%). No effect of ACPA (1 µg/mL) was seen in any of the investigated cells (0/24 cells, figure 4C, D).
APCA bind to CD68+ cells in vivo and in vitro, and induce CXCL1 release
To determine the cellular targets of ACPA, we performed immunohistochemical labelling of sections from mouse joints and bone. This revealed that ACPA, but not FT control, bind CD68+ cells, which based on CD68 immunoreactivity, multinucleated morphology and proximity to mineralised bone23 ,24 most likely are osteoclasts, and cells with the characteristics of osteoclast precursor cells in the bone marrow (see figure 5A and online supplementary figure S3A). ACPA did not label synoviocytes, chondrocytes, osteocytes or PECAM-1+ endothelial cells (see figure 5B and online supplementary figure S3B). Interestingly, some ACPA+ cells were located in very close proximity to CGRP+ sensory fibres in the bone marrow (figure 5C). ACPA immunoreactivity was observed on the cell surface of cultured non-permeabilised CD68+ precursor cells and multinucleated osteoclasts (figure 5D) indicating that the ACPA epitope(s) are expressed on the plasma membrane.
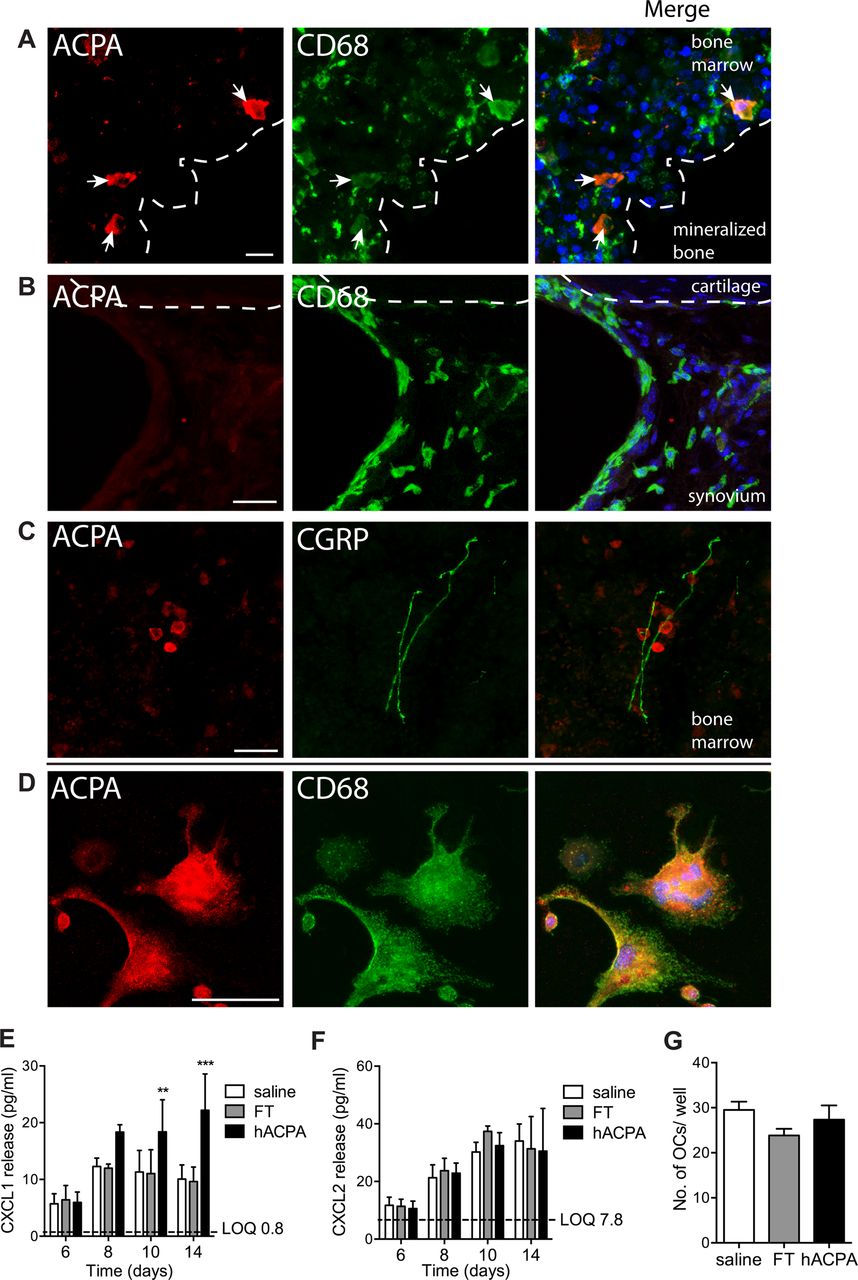
Binding of ACPA in tibial bone marrow and effect of ACPA on cultured osteoclasts. Colocalisation of ACPA: marker for macrophage/osteoclasts (CD68) in subchondral bone (A) and synovia (B), and marker for sensory nerve fibres (CGRP) in tibial bone marrow (C). ACPA and CD68 binding in cultured mouse bone marrow without permeabilisation of the plasma membrane (D). CXCL1 (E) and CXCL2 (F) levels in the supernatant of cultured mouse osteoclasts after stimulation with human ACPA (1 μg/mL), FT (1 μg/mL) or saline (n=6 mice/group). Three different cohorts of littermates were used (E–F). Number of osteoclasts per well at the end of experiment day 14 (G). Data are presented as mean±SEM. **p<0.01 and ***p<0.001 are compared with saline. Scale bar is 25 μm. ACPA, anti-citrullinated protein antibodies; FT, flow through.
In parallel work in one of our laboratories, we found that interleukin (IL) 8 is released by human osteoclasts in response to ACPA stimulation.25 We tested if ACPA drive release of IL-8 analogues also from cultured mouse osteoclasts by adding ACPA or FT on day 6, when multinucleated cells (osteoclasts) had started to form. We found that ACPA, but not FT, induced a significant release of CXCL1 (figure 5E) within 4 days in the presence of ACPA, while CXCL2 levels (figure 5F) and number of osteoclasts (figure 5G) did not change.
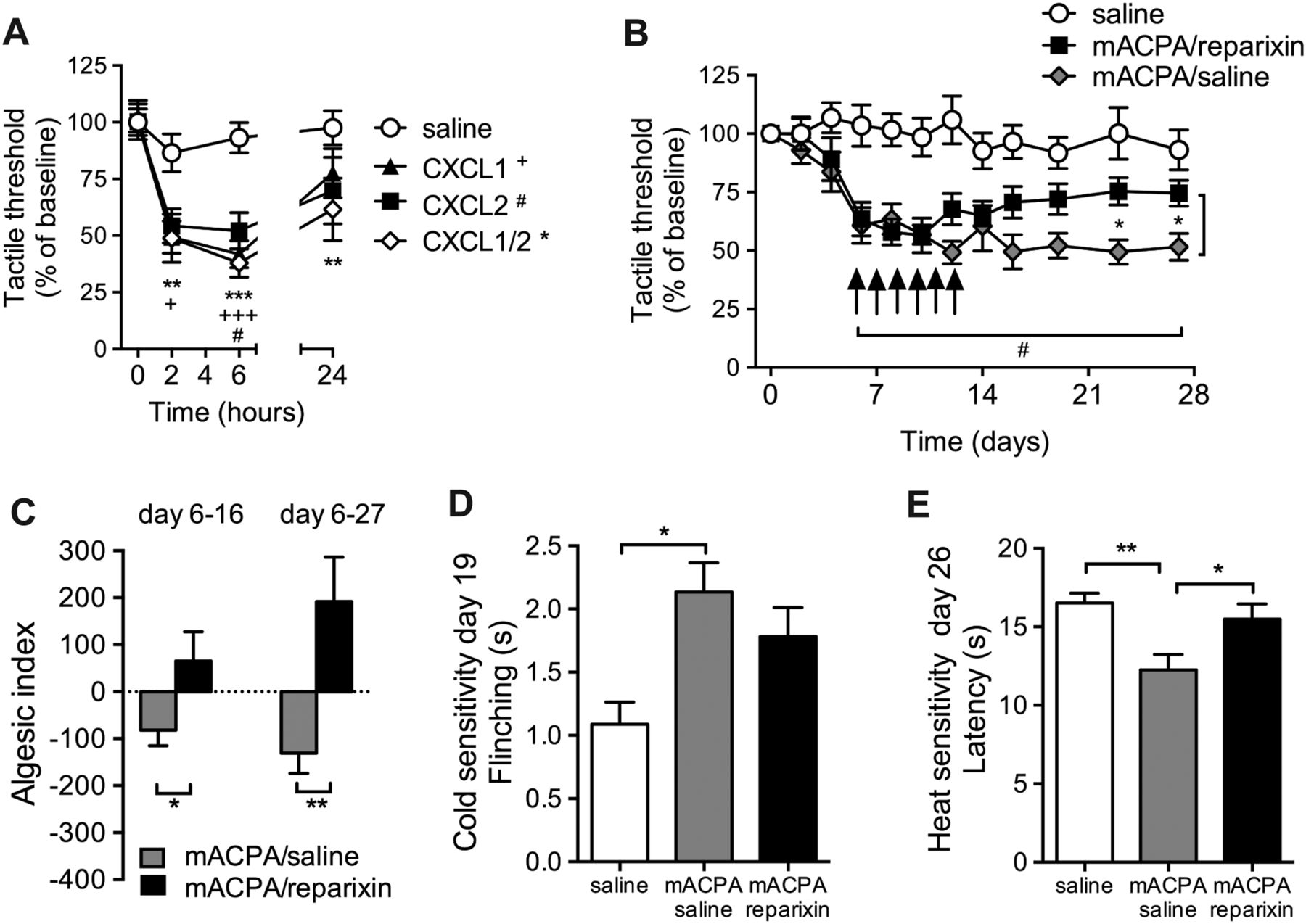
Pain-like behaviour is dependent on CXCL1/2
Injection of CXCL1 and/or CXCL2 into the ankle joint of mice produced a rapid onset of mechanical sensitivity in the ipsilateral paw, lasting at least 24 h (figure 6A). To examine the functional coupling between ACPA, CXCL1 release and nociception in vivo, mice with mechanical and thermal hypersensitivity induced by intravenous injection of monoclonal ACPA (D10 and B2) were treated with the CXCR1/2 receptor antagonist reparixin. Six consecutive days of reparixin injections (starting day 6) partially reversed mechanical hypersensitivity compared with saline controls between day 16 and the end of the study (day 28) (figure 6B, C) and sensitivity to heat and cold assessed on days 26 and 19, respectively (figure 6D, E). Reparixin treatment or injection of control antibody did not alter tactile thresholds in naive mice (online supplementary figure 4A-B).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of reparixin on ACPA-induced hypersensitivity. Mechanical hypersensitivity after injection of CXCL1 (30 ng, n=7), CXCL2 (30 ng, n=7) or mixed CXCL1/2 (15 ng each, n=10) or saline (n=20) into the ankle joint (A). Mechanical sensitivity after intravenous injection of mouse monoclonal ACPA D10 and B2 (1 mg each, n=18) or saline (n=9) and treatment with reparixin (30 mg/kg/day, s.c., n=9) or saline (n=9) for 6 days, starting on day 6 (B). Hyperalgesic index comparing area under the curve for reparixin-treated or saline-treated mice from day 6 (C). Cold (D) and heat (E) sensitivity were tested on days 19 and 26, respectively. Results are from two separate experiments. Statistical significance (two-way analysis of variance (ANOVA)) between mACPA/saline and saline is marked by # and difference between mACPA/saline and mACPA/reparixin is marked with * (A). Mechanical sensitivity of mice injected with either saline (n=5) or reparixin (30 mg/kg/day, s.c., n=5) on day 6–12 (E). Data are presented as mean±SEM. * or #p<0.05, **p<0.01 and ***p<0.001 are compared with saline. ACPA, anti-citrullinated protein antibodies.
Discussion
Here we provide evidence that ACPA, a family of autoantibodies that is common in patients with RA, induces pronounced pain-like behaviour in mice. While we did not identify a direct action of ACPA on sensory neurons, we found that (a) ACPA binds CD68+ osteoclasts in the bone marrow and induces CXCL1/2 expression in the joints and CXCL1 release, (b) intra-articular injection of CXCL1/2 evokes pain-like behaviour and (c) blockade of the chemokine receptors for CXCL1/2 attenuates ACPA-induced hypersensitivity. Thus, our study shows that ACPA-induced nociception is mediated via a mechanism that is dependent on IL-8 release. Importantly, this effect is specific for ACPA, as antibodies isolated from patients who were ACPA- and the FT containing the non-ACPA antibodies from patients who were ACPA+ did not increase mechanical or thermal sensitivity or alter locomotor behaviour in mice. Additionally, murinised monoclonal ACPA induced similar pain-like behaviour in mice as the human ACPA, which excludes potential bias of pronociceptive mechanisms being initiated by immune reactions against human proteins.
We did not observe any visual or histological signs of inflammation in the joints after systemic administration of ACPA. Of the different inflammation-associated factors that were assessed, only CXCL1 and CXCL2 mRNA levels were increased, further strengthening the concept that ACPA-induced pain occurs without the presence of classic signs of inflammation. Thus, our data highlight a potential role of ACPA in the type of joint pain that precedes development of RA and/or persists despite medical control of the disease activity. In the current studies we used polyclonal human antibodies and mouse monoclonal antibodies, which are specific for citrullinated peptides, but which among them have different reactivity patterns for different citrullinated epitopes on different potential target molecules. This variation in reactivity is typical for most of the human B cell and plasma cell derived monoclonals that we have generated so far from RA joints.18 Since both different batches of polyclonal ACPA and the different monoclonal ACPA varied in their nociceptive potency, it is plausible that certain specific citrullinated epitopes are critical for the induction of pain-like behaviour. This may be part of the reason for why subgroups and not all ACPA+ individuals develop arthralgia. Mapping the nociceptive effect of antibodies with defined fine-specifies will therefore be important in future detailed studies on the molecular mechanisms involved in ACPA-induced pain.
We found that ACPA binds surface epitopes on CD68+ precursor cells and osteoclast in the mouse bone marrow, subchondral bone and growth plate, but did not detect any ACPA-specific immunoreactivity in other regions of the bone and joints, including the synovium. In parallel work,25 we show that the same monoclonal ACPAs that induced pain-like behaviour also show plasma membrane immunoreactivity in cultured human CD68+ osteoclasts and increase their activity in vitro and reduce bone volume and trabeculations in vivo in mice after long-term (4 weeks) exposure to these antibodies. Furthermore, not all ACPA are pronociceptive; the monoclonal ACPA (C7) that lacked pronociceptive properties also failed to induce osteoclast activation in vitro. Thus, there is a correlation between ACPA-mediated effects on osteoclasts and bone metabolism on one hand and activation of the sensory nervous system on the other. Furthermore, while ACPA stimulation evoked IL-8 release from both mouse and human osteoclasts in vitro,25 ACPA failed to induce inward currents and Ca2+ fluxes in cultured mouse DRG neurons, implicating that ACPA does not directly modify neuronal excitability, at least not during the conditions used in the present study. These observations lead to the hypothesis that ACPA induce hypersensitivity via IL-8 release from CD68+ cells in the bone marrow.
The CXCL (IL-8) class of chemokines has been reported to induce pain-like behaviour when injected into peripheral tissues26 ,27 or into the spinal fluid28 of rodents, acting via CXCR2 expressed on peripheral and central nociceptive neurons.28–30 Furthermore, CXCL1 increases voltage-gated Na+ and K+ currents and the function of TRPV1 in murine peripheral DRG neurons, contributing to heightened sensitisation and excitability of the peripheral sensory neurons.31 ,32 We detected increased levels of CXCL1 and CXCL2 mRNA in the joints of ACPA-injected mice and injection of CXCL1/2 as a mix, or one by one, into the ankle joint induced mechanical hypersensitivity, confirming a direct involvement of CXCL1/2 in nociception. Since ACPA stimulation did not evoke release of other pain-associated cytokines such as TNF, IL-6 and IL-1β,25 but caused release of CXCL1/IL-8 in both mouse and human osteoclasts cultures, osteoclasts and CXCL1 provide an intriguing link between ACPA administration and development of pain-like behaviour. We found ACPA immunoreactive cells in the bone marrow located in close proximity to CGRP+ sensory nerves, which provides an intriguing histological link that supports the presence of an interaction between osteoclasts and pain fibres. In addition, blocking the action of CXCL1/2 using reparixin, a CXCL1/2 receptor antagonist in clinical development, reversed ACPA-induced hypersensitivity, pointing to a direct causal relationship between ACPA, CXCL1/2 and pain.
To date citrullinated vimentin14 and possibly α enolase25 has been identified as critical epitopes for ACPA-mediated effects on osteoclasts, but other epitopes may also be important. Of note, the Fab part of the ACPA induce osteoclast activation,25 and thus the reported mechanism is not likely to be solely dependent on Fc-part of the antibodies, although a contribution of activation of Fcγ receptors on osteoclasts cannot be excluded.33 ,34 The precise mechanism by which ACPA activate osteoclasts and induce IL-8 release warrants further studies. Our data suggest that CXCL1/IL-8 released from osteoclasts act on nearby sensory neurons through the receptor CXCR2. As we did not observe pain-like behaviour until 2 days after injection of ACPA, it is likely that this process requires ACPA-driven osteoclast differentiation followed by sufficient release of IL-8 to cause peripheral neuronal sensitisation.
In conclusion, our present findings open the possibility that the arthralgia that often precedes the onset of RA may be a direct consequence of the presence of certain ACPA, rather than an unspecific symptom unrelated to the pathogenesis of RA. This insight should dramatically alter our approach to diagnosing as well as treating ACPA+ arthralgia, and may indicate new potential targets for the prevention of development of clinical signs of RA in this early phase of disease development. Such targets would obviously include both IL-8 and associated receptors and factors in osteoclasts that contribute to the ACPA-induced production of IL-8. Our findings may also provide a possible explanation to the remaining pain in some patients with ACPA+ RA who have been successfully treated for their inflammation; levels of ACPA do normally persist also after successful treatment of inflammation.35 ,36
Acknowledgments
Thanks to Stephen Rapecki (UCB Celltech, Slough, England, UK) for murinisation of the human monoclonal ACPAs, to Heidi Wähämaa for purification of human antibodies and to Marie Björnholm and Anna Krook for technical advice on the Oxymax/Comprehensive Lab Monitoring System.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
DBB and CFC contributed equally.
Correction notice This article has been corrected since it published Online First and in print. The title has been corrected to: Autoantibodies to citrullinated proteins may induce joint pain independent of inflammation.
Contributors GW and CIS designed experiments, analysed the data and wrote the manuscript along with P-JJ, AIC, KL, RH, VM and LK. GW conducted histology and qPCR, and together with JS, KR, JK and KS behavioural experiments. GW and JMJ-A developed protocols and performed ACPA IHC in joint and bone. DBB and ABF cultured DRG neurons and performed in vitro DRG experiments. CF-C purified human antibodies and performed western blots. AK made osteoclast cultures and associated experiments. AF measured chemokines in supernatants. KSN and RH supplied mice and CAIA antibodies. KA and VM produced mACPA. GW, CIS, DBB, CF-C, JK, P-JJ, AIC, KL, KR, VM, KSN, RH and LK discussed and developed the concept.
Funding This work was supported by the Swedish Research Council, Swedish Foundation for Strategic Research, Ragnar Söderberg Foundation, Knut and Alice Wallenberg Foundation, the Karolinska Institutet Foundation, Foundation Konung Gustaf V:s 80-årsfond, the European Research Council and the IMI programme BTCure (115142-2).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Basic and translational research
- Miscellaneous