Article Text

Abstract
Objective Gout, caused by hyperuricaemia, is a multifactorial disease. Although genome-wide association studies (GWASs) of gout have been reported, they included self-reported gout cases in which clinical information was insufficient. Therefore, the relationship between genetic variation and clinical subtypes of gout remains unclear. Here, we first performed a GWAS of clinically defined gout cases only.
Methods A GWAS was conducted with 945 patients with clinically defined gout and 1213 controls in a Japanese male population, followed by replication study of 1048 clinically defined cases and 1334 controls.
Results Five gout susceptibility loci were identified at the genome-wide significance level (p<5.0×10−8), which contained well-known urate transporter genes (ABCG2 and SLC2A9) and additional genes: rs1260326 (p=1.9×10−12; OR=1.36) of GCKR (a gene for glucose and lipid metabolism), rs2188380 (p=1.6×10−23; OR=1.75) of MYL2-CUX2 (genes associated with cholesterol and diabetes mellitus) and rs4073582 (p=6.4×10−9; OR=1.66) of CNIH-2 (a gene for regulation of glutamate signalling). The latter two are identified as novel gout loci. Furthermore, among the identified single-nucleotide polymorphisms (SNPs), we demonstrated that the SNPs of ABCG2 and SLC2A9 were differentially associated with types of gout and clinical parameters underlying specific subtypes (renal underexcretion type and renal overload type). The effect of the risk allele of each SNP on clinical parameters showed significant linear relationships with the ratio of the case–control ORs for two distinct types of gout (r=0.96 [p=4.8×10−4] for urate clearance and r=0.96 [p=5.0×10−4] for urinary urate excretion).
Conclusions Our findings provide clues to better understand the pathogenesis of gout and will be useful for development of companion diagnostics.
- Gout
- Arthritis
- Gene Polymorphism
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Gout is a common disease caused by deposition of monosodium urate (MSU) crystal due to hyperuricaemia.1 Humans have long suffered from gout as reported by Hippocrates 2500 years ago.2 There have been many famous patients with gout such as Sir Isaac Newton3 in the more recent past, and the numbers are still growing. From the pathophysiological point of view, gout can be classified into the renal underexcretion (RUE) type, the renal overload (ROL) type and the combined type based on clinical parameters4 (see online supplementary figure S1).
So far the genome-wide association studies (GWASs) of serum uric acid (SUA) level5–16 have identified a number of genetic loci including SLC2A9 (also known as GLUT9) and ABCG2 (also known as BCRP), and subsequent genetic and functional studies have revealed the biological and pathophysiological significance of ABCG2.4 ,17 ,18 Previous GWASs of gout reported a significant association with single-nucleotide polymorphisms (SNPs) of ABCG2, SLC2A9 with European ancestries,14 ,15 and of ALDH16A1 with Icelanders,14 while another study with African-American and European ancestries reported no significantly associated SNPs of gout.13 All of these studies were, however, performed with cases including self-reported patients with gout, in which clinical information was insufficient. Therefore, the relation to genetic heterogeneity underlying gout subtypes is also unclear. To better understand its genetic basis, we first performed a GWAS of clinically defined gout cases only. We then investigated the relationship between genetic variation and clinical types of gout.
Methods
Subjects
In the present study, we avoided use of self-reported gout cases and collected only clinically defined gout cases. All gout cases were clinically diagnosed as primary gout according to the criteria established by the American College of Rheumatology.19 All patients were assigned from among the Japanese male outpatients at the gout clinics of Midorigaoka Hospital (Osaka, Japan), Kyoto Industrial Health Association (Kyoto, Japan) or Ryougoku East Gate Clinic (Tokyo, Japan). Patients with inherited metabolism disorders including Lesch–Nyhan syndrome were excluded. Finally, 1994 male gout cases were registered as valid case participants. As controls, 2547 individuals were assigned from among Japanese men with normal SUA level (≤7.0 mg/dL) and no gout history, who were obtained from BioBank Japan11 ,20 and Japan Multi-Institutional Collaborative Cohort Study (J-MICC Study).21
Genotyping and quality control
Genome-wide genotyping was performed with Illumina HumanOmniExpress v1.0 (Illumina) in 946 cases and 1213 controls. Detailed methods of genotyping and quality control are shown in the online supplementary methods and figure S2. Finally, 570 442 SNPs passed filters for 945 cases and 1213 controls.
In total, 123 SNPs passing the significance threshold at p<1.0×10−5 in the GWAS stage were used for subsequent analyses. Among these SNPs, we examined their linkage disequilibrium (LD) and selected 16 SNPs for replication study (see online supplementary methods). These 16 SNPs were then genotyped by an allelic discrimination assay (Custom TaqMan Assay and By-Design, Applied Biosystems) with a LightCycler 480 (Roche Diagnostics).18 After quality control, subsequent statistical analysis was performed with 1048 cases and 1334 controls.
Statistical analyses for GWAS
We conducted an association analysis using a 2×2 contingency table based on the allele frequency, and p value of association was assessed by χ2 test. The quantile–quantile plot and the genomic inflation factor were used to assess the presence of systematic bias in the test statistics due to potential population stratification (see online supplementary methods and figure S3).
We then combined results from the GWAS and replication stages by meta-analysis.22 The inverse-variance fixed-effects model meta-analysis was used for estimating summary OR. Cochran's Q test23 and I2 statistic24 ,25 were examined to assess heterogeneity in ORs between GWAS and replication study. If heterogeneity was present by the statistical test (phet<0.05) or measurement (I2>50%), we implemented DerSimonian and Laird random-effects model meta-analysis.26 All the meta-analyses were performed using the STATA V.11.0. Genome-wide significance threshold was set to be α=5.0×10−8 to claim evidence of a significant association. Detailed methods of imputation and per cent variance are shown in the online supplementary methods.
Subtype analyses
Gout contains two distinct types, ‘ROL’ type and ‘RUE’ type. The ROL type was defined when urinary urate excretion (UUE) was over 25.0 mg/h/1.73 m2 (600 mg/day/1.73 m2)4 ,27–29 and their urate clearance (urate clearance/creatinine clearance ratio, FEUA) was 5.5% or over. Also, the RUE type was determined when UUE was 25.0 mg/h/1.73 m2 or under and FEUA was under 5.5%.4 ,30 ,31 Detailed methods of subtype analyses are described in the online supplementary methods.
Results
Genome-wide association study
Clinical characteristics of participants in this study are shown in online supplementary tables S1–S3. GWAS with 945 clinically defined gout cases and 1213 controls identified SNPs in three loci showing evidence of associations at the genome-wide significance level (p<5.0×10−8): rs2728125 of ABCG2 (p=1.5×10−27; OR=2.05), rs3775948 of SLC2A9 (p=6.7×10−15; OR=1.64) and rs2188380 of MYL2-CUX2 (p=5.7×10−13; OR=1.78, figures 1 and 2, table 1 and online supplementary figure S4).
Five SNPs showing significant association at genome-wide significance level and one suggestive SNP

Manhattan plot of a genome-wide association analysis of gout. X-axis shows chromosomal positions. Y-axis shows -log10 p values. The upper and lower dotted lines indicate the genome-wide significance threshold (p=5.0×10−8) and the cut-off level for selecting single-nucleotide polymorphisms for replication study (p=1.0×10−5), respectively.
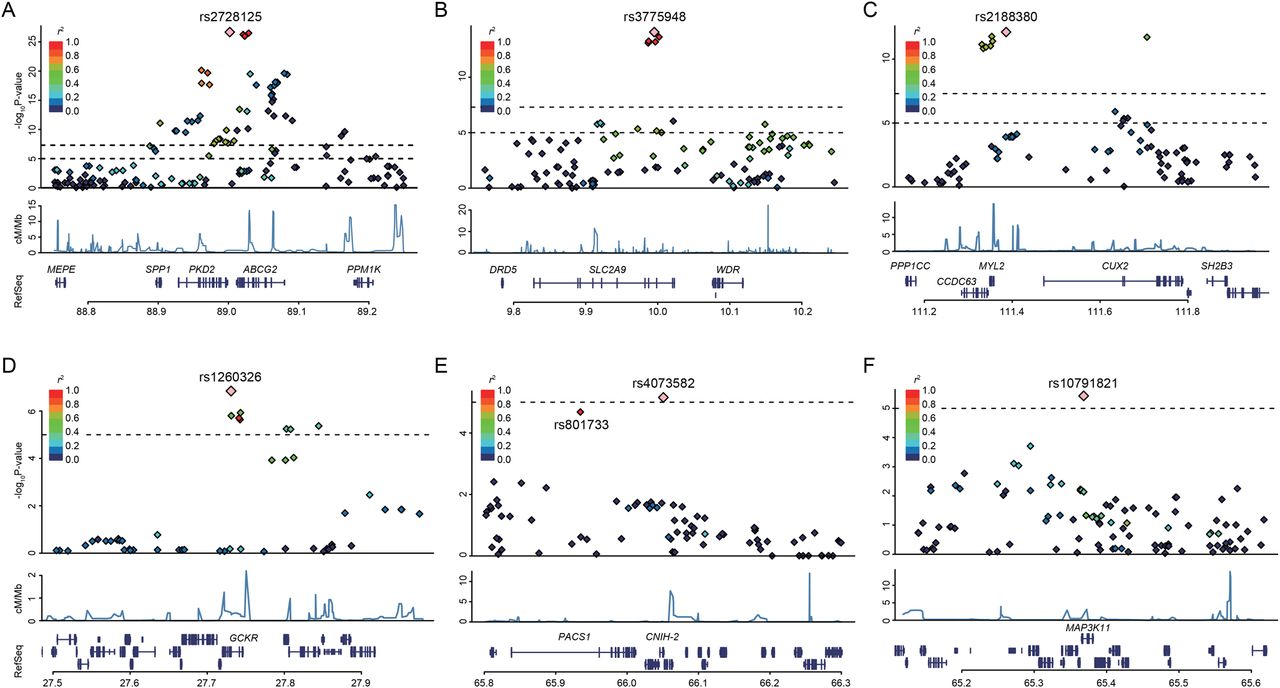
Regional association plots for six discovered loci of gout. Five regions exceeding the genome-wide significance level (A–E) and one region showing a suggestive association (F). The highest association signal in each panel is located on ABCG2 (A), SLC2A9 (B), MYL2-CUX2 (C), GCKR (D), CNIH-2 (E) and MAP3K11 (F). Region within 250 kb from single-nucleotide polymorphism (SNP) showing lowest p value is displayed. (Top panel) Plots of -log10 p values for the test of SNP association with gout in the genome-wide association study stage. SNP showing the lowest p value is depicted as a pink diamond. Other SNPs are colour-coded according to the extent of linkage disequilibrium (measured in r2) with SNP showing the lowest p value. (Middle panel) Recombination rates (centimorgans per Mb) estimated from HapMap Phase II data are plotted. (Bottom panel) RefSeq genes. Genomic coordinates are based on Genomic Reference Consortium GRCh37.
Replication study was conducted with 1048 cases and 1334 controls. As a result, the three SNPs surpassing the genome-wide significance threshold in the GWAS stage were successfully replicated; rs2728125 (p=8.3×10−29; OR=2.03), rs3775948 (p=7.6×10−14; OR=1.57) and rs2188380 (p=2.0×10−12; OR=1.73). Additionally, two SNPs showed significant associations at p<3.1×10−3 (=0.05/16) with Bonferroni correction; rs1260326 of GCKR (p=2.8×10−6; OR=1.32) and rs4073582 of CNIH-2 (p=1.6×10−4; OR=1.55) as shown in table 1 and online supplementary table S4.
All five SNPs that showed significant associations in the replication study achieved genome-wide significance in the meta-analysis of GWAS and replication study (table 1): rs2728125 (pmeta=7.2×10−54; OR=2.04), rs3775948 (pmeta=5.5×10−27; OR=1.61), rs2188380 (pmeta=1.6×10−23; OR=1.75), rs1260326 (pmeta=1.9×10−12; OR=1.36) and rs4073582 (pmeta=6.4×10−9; OR=1.66). In addition, an intronic SNP of MAP3K11 (rs10791821) showed a suggestive level of association (pmeta=1.0×10−7; OR=1.57). There was >80% power to detect a risk variant for OR=1.6 at the genome-wide significance level (p<5.0×10−8) for an SNP with a minor allele frequency of 0.35 (see online supplementary table S5). Imputation was also performed with the GWAS genotyping data for 1 Mb across the identified SNPs of novel loci (rs2188380 of MYL2-CUX2, rs1260326 of GCKR, rs4073582 of CNIH-2 and rs10791821 of MAP3K11). SNPs that passed the significant threshold of GWAS stage (p<1.0×10−5) in this imputation are shown in online supplementary table S6A–D.
Two dysfunctional SNPs of ABCG2
We previously demonstrated that two dysfunctional SNPs of ABCG2, rs72552713 (Gln126Ter) and rs2231142 (Gln141Lys), were located on different haplotypes4 ,18 and strongly associated with hyperuricaemia and gout.4 ,18 ,32 Therefore, we additionally performed genotyping of these two SNPs by an allelic discrimination assay because SNPs are not on Illumina HumanOmniExpress V.1.0 (Illumina). SNP showing the highest significance in the present GWAS (rs2728125) was in strong LD with rs2231142 (r2=0.76) but not in LD with rs72552713 (r2=0.03). A multivariate logistic regression analysis including these three SNPs of ABCG2 showed that rs2728125 no longer had a significant association (p=0.19), but rs72552713 and rs2231142, that is, two non-synonymous SNPs, remained highly significant (see online supplementary table S7A, B), indicating that rs2728125 was merely a surrogate marker for rs2231142. Therefore, we used these two non-synonymous variants for subsequent analyses.
Cumulative effect of risk alleles for gout
Accumulation of the number of risk alleles of the gout-associated SNPs (rs3775948, rs2188380, rs1260326, rs4073582, rs72552713 and rs2231142) increased the probability of gout logarithmically. When setting the reference category as having four or fewer risk alleles, ORs for having 5, 6, 7, 8 and 9 or more risk alleles were 1.79 (p=3.5×10−3), 3.16 (p=2.3×10−10), 5.10 (p=9.7×10−21), 10.1 (p=5.3×10−39) and 18.6 (p=3.6×10−45), respectively (see online supplementary figure S5 and table S8).
Subtype analysis of gout
We examined type-specific ORs and the case–subtype heterogeneity test.33 The subgroup analysis (table 2) showed that the associations of two non-synonymous SNPs of ABCG2 (rs72552713 and rs2231142) were stronger for the ROL type (ORs=4.35 and 3.37, respectively) than for the RUE type (ORs=1.28 and 1.88, respectively). The differences in ORs between the gout types were highly significant (p=2.4×10−5 and 1.0×10−7, respectively). The association of rs3775948 of SLC2A9 was stronger for the RUE type (OR=1.94) than for the ROL type (OR=1.38). The case–subtype heterogeneity test showed a significant difference in ORs (p=2.7×10−4). The other SNPs evidenced no significant differences. Then, associations between SNPs and clinical parameters (FEUA and UUE) were assessed. Only SNPs that showed a significant difference in ORs between different gout types were significantly associated with FEUA and UUE (table 2 and online supplementary figure S6, table S9); the gout risk alleles of ABCG2 and SLC2A9 were associated with increased and decreased levels of these parameters, respectively. The effect of the risk allele of each SNP on clinical parameters showed significant linear relationships with OR in the case–subtype heterogeneity test, which was an estimate of the ratio of the case–control ORs for the gout types (r=0.96 [p=4.8×10−4] for FEUA and r=0.96 [p=5.0×10−4] for UUE) (figure 3).
Associations of seven SNPs with gout types
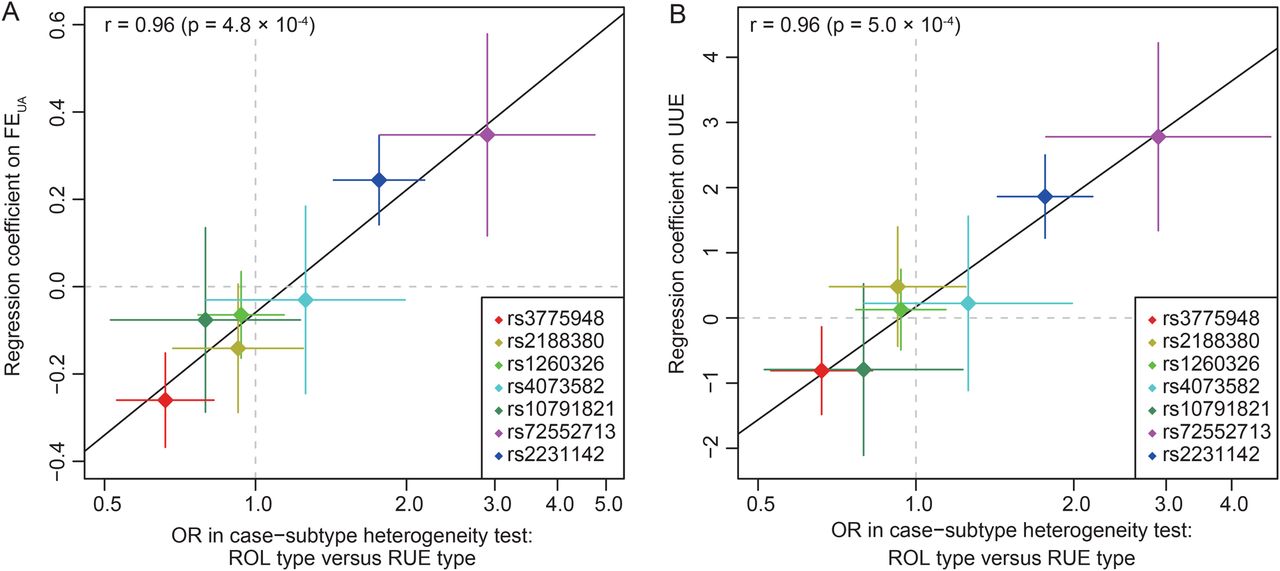
Relationships between effects of risk alleles on clinical parameters and ORs in case–subtype heterogeneity test. (A) FEUA and (B) urinary urate excretion (UUE). The seven single-nucleotide polymorphisms (SNPs) listed in table 2 were examined. OR in case–subtype heterogeneity test is an estimate of the ratio of the case–control OR for the renal overload (ROL) type to that for the renal underexcretion (RUE) type. If an SNP has a stronger effect for the ROL type than for the RUE type, it takes a value >1. Diamonds and lines represent point estimates and their 95% CIs. Pearson's correlation coefficient (r) between the effect on clinical parameters and natural logarithm of OR in case–subtype heterogeneity test and its significance were examined. FEUA, fractional excretion of urate clearance.
Discussion
Through the GWAS with clinically defined cases, we identified five gout-associated loci that showed different association patterns in subtype analysis. Previous GWASs of SUA5–16 showed genome-wide significant associations with ABCG2, SLC2A9 and GCKR. These genes were also reported to have significant associations with gout as a consequence of hyperuricaemia.13–15 The present study revealed for the first time that three loci (GCKR, MYL2-CUX2 and CNIH-2) were associated with gout at the genome-wide significance level. In particular, MYL2-CUX2 and CNIH-2 are novel loci for gout.
The total variance explained by the seven SNPs was estimated to be 9.0% (see online supplementary methods): three SNPs of well-known urate transporter genes (SLC2A9 and ABCG2) with large effects accounted for 6.9%, and the four SNPs identified in this GWAS with modest effects explained 2.1%. Additional discoveries of unidentified genetic variants by performing a meta-analysis of GWAS data sets will improve the explained genetic variation of gout.
ABCG2 and SLC2A9 are well-known urate transporter genes for urate excretion17 ,18 and renal urate reabsorption,34 ,35 respectively. ABCG2 is identified to have an association with SUA levels by recent GWASs.9–16 Subsequent genetic and functional analysis17 ,18 revealed that ABCG2 is a high-capacity urate exporter and shows the reduced transport of urate by a common half-functional variant, rs2231142 (Gln141Lys). We also demonstrated that common dysfunctional genotype combinations of ABCG2 gene (non-functional rs72552713 [Gln126Ter] and rs2231142) are a major cause of hyperuricaemia and gout,18 especially for early-onset gout.32 We earlier found that the risk alleles of these two SNPs reside on different haplotypes,4 ,18 indicating independent risks of gout. Recently, these dysfunctional SNPs were revealed to decrease extrarenal (intestinal) urate excretion and to cause ROL hyperuricaemia,4 through studies with hyperuricaemic patients4 and Abcg2-knockout mice.4 ,36 This is consistent with the fact that ABCG2 exporter is expressed on the apical membrane in several tissues, including intestine37 and kidney,38 which have urate-excreting functions in humans.
SLC2A9 is a member of the glucose transporter (GLUT) family. SLC2A9 was found to transport urate,7 ,34 and several GWAS have demonstrated an association of SLC2A9 with SUA levels.5–16 SLC2A9 has two isoforms, GLUT9L (long isoform) and GLUT9S (short isoform),34 and is highly expressed in the kidney proximal tubules in humans.39 Genetic and functional analysis34 ,35 with patients with renal hypouricaemia (RHUC) revealed that RHUC is caused by dysfunctional mutations in SLC2A9, which decrease urate reabsorption in the renal proximal tubules. For example, non-functional mutations of either GLUT9L (Arg198Cys and Arg380Trp) or GLUT9S (Arg169Cys and Arg351Trp, corresponding to Arg198Cys and Arg380Trp in GLUT9L), which were found from patients with RHUC, dramatically reduced the urate transport activity.34 Therefore, SLC2A9 plays an important role in renal urate reabsorption.34 Thus, SLC2A9 is a causative gene for RHUC type 2,34 ,40 which was confirmed by the report of homozygous mutations in patients with RHUC type 2.35 In our subtype analysis, OR of RUE type was higher than that of ROL type (OR=1.94 and 1.38, respectively, table 2), which is compatible with the fact that SLC2A9 is a transporter for urate reabsorption in human kidney.
Glucokinase regulatory protein (GCKR) controls the activity of glucokinase, which is a major glucose sensor for insulin secretion. GCKR regulates the first step of glycolysis, the phosphorylation of glucose to glucose-6-phosphate.41 ,42 Glucokinase activity is controlled by GCKR, which binds to glucokinase and suppresses its function in the postabsorptive phase. On the other hand, this binding is loosened in the postprandial phase, so that glucokinase could adopt the glycolysis.43 So far, the gout risk allele of rs1260326 (Leu446Pro) has been reported to be associated with lower fasting glucose levels, and inversely, higher levels of triglyceride43–45 and SUA.10 ,12 ,15 An association of GCKR with dyslipidaemia has also been reported.46
MYL2 encodes a regulatory light chain associated with cardiac myosin β (or slow) heavy chain. MYL2 mutations are associated with mid-left ventricular-type hypertrophic cardiomyopathy. In addition, its association with high-density lipoprotein cholesterol metabolism was previously reported.47 CUX2 regulates cell-cycle progression48 and plays important roles in neural progenitor development in the central nervous system.48 ,49 Its association with type 1 diabetes has also been reported.50 Thus, rs2188380 of MYL2-CUX2 showed an association with gout because MYL2 and CUX2 might influence such metabolic pathways. Rs2188380 locates near rs653178 of ATXN2 (see online supplementary figure S7), which was reported by Köttgen et al15 to have an association with SUA. Rs653178 is, however, monomorphic in the Japanese population of the HapMap project,51 and we also confirmed it in our samples by genotyping >250 replication cases. Conversely, rs2188380 of MYL2-CUX2 is monomorphic in European and African populations,51 while rs2188380 is a common variant in the Japanese population (table 1). Therefore, this SNP was identified as a novel locus of gout in the present study. The differences in study populations could be one of the reasons why rs2188380 was not found in a large European-driven GWAS on urate and gout.15 Further analyses including fine mapping and functional analysis are required in this region.
CNIH-2 regulates the function of glutamate receptors of the AMPA-subtype assembly at the cell surface of various neurons and glial cells.52 ,53 CNIH-2 modulates AMPA receptor gating by increasing its cell surface expression. The newly identified rs4073582 of CNIH-2 was in strong LD with rs801733 in PACS1 (r2=0.97, figure 2E and see online supplementary figure S4E), which is reported to be associated with severe obesity.54 Accordingly, PACS1 can also be a good candidate for a gout susceptibility gene. Additional genetic dissection and functional analysis will be needed to determine whether these genes or others could play roles with true causality at this locus. Since Okada et al16 previously reported the association between SUA and rs504915 of NRXN2, which is near CNIH-2 and MAP3K11, we examined their relationships. They are not in strong LD (see online supplementary table S10), and the association of rs4073582 and rs10791821 remained significant after adjustment with rs504915 (see online supplementary table S11). Therefore, rs4073582 of CNIH-2, rs10791821 of MAP3K11 and rs504915 of NRXN2 are revealed to be independent of each other.
MAP3K11, also known as mixed lineage kinase 3 (MLK3), is a MAP kinase member and plays a significant role in the activation of c-Jun N-terminal kinase (JNK), a stress-activated protein kinase.55 Signalling from the small GTP-binding proteins Rac1 and Cdc42 induces MLK3 to activate the MEKK-SEK-JNK kinase cascade. Interestingly, the JNK pathway is activated when monocytes/macrophages phagocytose MSU crystals,56 which cause gouty arthritis. The SNP rs10791821 of MAP3K11 has been associated with the expression level of MAP3K11 in monocytes,57 and therefore, is likely to be a regulatory SNP. However, further study is required to confirm precise involvement of MAP3K11 in the development of gout.
Other genes (CCDC63, C2orf16, ZNF512, RAB1B, EHBP1L1 and KCNK7) near each of the novel loci, which are found by imputation analysis (see online supplementary table S6A–D), could also be candidate genes of gout, and further studies including functional analyses are warranted.
Most of the gout-related genes are also associated with SUA.15 In the present study design, to identify novel gout risk loci, clinically defined gout and normouricaemic controls were recruited. Therefore, further investigations with different study designs will be needed to identify gout loci associated with crystal deposition and inflammation.
We further investigated the cumulative effect of risk alleles of the five significant loci (ABCG2, SLC2A9, MYL2-CUX2, GCKR and CNIH-2) on gout risk. The result showed that individuals with five or more risk alleles had a higher risk for gout compared with those having four or fewer risk alleles. The more risk alleles in an individual, the higher became the risk of gout.
Furthermore, the relationship between genetic variation and clinical types of gout was investigated. The results of subtype analyses (table 2, figure 3 and online supplementary figure S6, table S9) indicate that the alleles closely associated with the risk of specific gout type represented differential effects on clinical parameters (FEUA and UUE). This allows the estimation of disturbed urate excretion pathways. An increase of FEUA and UUE by the risk alleles of ABCG2 leads to the overloading effect on renal urate excretion and causes the ROL-type gout (figure 4A). These estimations are consistent with our previous finding obtained from Abcg2-knockout mouse models and hyperuricaemic patients.4 In contrast, the reduction of FEUA and UUE by the risk allele of SLC2A9 reflects a decreased renal urate excretion, thereby increasing the risk of the RUE-type gout (figure 4B). The present study demonstrated that the combination of GWAS of patients with clinically defined gout with actual clinical data is an effective method to analyse genetic heterogeneity among different types of gout.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Differential effects by risk allele on clinical parameters and gout. (A) The risk alleles of ABCG2 increase UUE and FEUA, which leads to the overloading effect on renal urate excretion and increases the risk of the ROL-type gout. Therefore, patients with risk alleles for the ROL-type gout would be given urate synthesis inhibitors. (B) The risk allele of SLC2A9 reduces UUE and FEUA, which reflects a decreased renal urate excretion, thereby increasing the risk of the RUE-type gout. Patients with risk alleles for the RUE-type gout would be administered uricosuric agents. FEUA, fractional excretion of urate clearance; ROL, renal overload; RUE, renal underexcretion; SUA, serum uric acid; UUE, urinary urate excretion.
In summary, we conducted the first GWAS using patients with clinically defined gout only and identified five loci containing two novel loci. Moreover, identified SNPs showed differential effects on different gout types and affected clinical parameters underlying specific types. Thus, genetic testing for gout may well be introduced into future companion diagnostics. For example, patients with risk alleles for ROL-type gout would be given urate synthesis inhibitors31 ,58 such as allopurinol and febuxostat, while patients with risk alleles for RUE-type gout would be administered uricosuric agents31 ,58 including benzbromarone and lesinurad, a selective uric acid reabsorption inhibitor that has just finished its phase III study.59 ,60 Exploring genetic heterogeneity among different gout types will deepen understanding of the aetiology of gout and serve to categorise patients for future personalised treatment.
Acknowledgments
We would like to thank all the participants involved in this study. We also thank members of the BioBank Japan Project and Japan Multi-Institutional Collaborative Cohort Study (J-MICC Study) Shizuoka Field for supporting the study. We are indebted to K. Gotanda, Y. Morimoto, N. Katsuta, Y. Utsumi, Y. Kato, H. Sasaki, Y. Takashima, J. Sato, H. Inoue, C. Okada, S. Takeuchi, N. Otani, S. Tomura (National Defense Medical College), T. Tamatsukuri (Jikei University School of Medicine), Y. Oikawa and K. Niwa (Toho University) for genetic analysis; S. Ushida (Ikagaku), H. Fujiwara (Midorigaoka Hospital), A. Hishida and K. Wakai (Nagoya University) for sample collection; M. Hosoyamada, S. Fujimori (Teikyo University), T. Shimizu, T. Sugiura (Kanazawa University), H. Sato (National Defense Medical College), K. Shimono (Toho University) and T. Makino (Nagoya City University) for helpful discussion.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
HM, KY, HNakaoka, AN and MS contributed equally.
Contributors HM, KY, HNakaoka, AN, MS, TC, II and NS conceived and designed the experiments. ATakahashi, TN, HNakashima, YT, TT, YS, HS, YKanai, TH and MK assisted with research design. HM, AN, MS, TC, ATokumasu, KIchida, HOoyama and TS collected and analysed clinical data of cases. ATakahashi, GY, RO, EM, MN, NH and MK collected and analysed clinical data of controls. HM, KY, AN, MS, TC, YT, ID, SS, JA, YKawamura, STerashige, HOgata, STatsukawa, YN and NS performed genetic analysis. HNakaoka, ATakahashi, TN, HNakashima and YS performed statistical analysis. HM, KY, HNakaoka, AN, MS, TC, ATakahashi, TN, HNakashima, MK, and NS analysed the data. TN, HNakashima, HOnoue, KIwaya, TI, TT, KInoue, YKato and II provided intellectual input and assisted with the preparation of the manuscript. HM, KY, HNakaoka, AN, MS, TC, ATakahashi and NS wrote the paper.
Funding This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan including the MEXT KAKENHI (Grant numbers 221S0002, 25293145, 22689021, 25670307), the Ministry of Health, Labour and Welfare of Japan, the Ministry of Defense of Japan, the Japan Society for the Promotion of Science, the Kawano Masanori Memorial Foundation for Promotion of Pediatrics and the Gout Research Foundation of Japan. The BioBank Japan Project and J-MICC Study (221S0001) were supported by MEXT of Japan.
Competing interests HM, TT and NS have a patent pending based on the work reported in this paper. Other authors have declared that no competing interests exist.
Patient consent Obtained.
Ethics approval All procedures involved in this study were approved by the institutional ethical committees of National Defense Medical College, Nagoya University and RIKEN, and all procedures involved were performed in accordance with the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.