Article Text

Abstract
Objectives Systemic immunological processes are profoundly shaped by the micro-environments where antigen recognition occurs. Identifying molecular signatures distinctive of such processes is pivotal to understand pathogenic immune responses and manipulate them for therapeutic purposes. Unfortunately, direct investigation of peripheral tissues, enriched in pathogenic T cells, is often impossible or imposingly invasive in humans. Conversely, blood is easily accessible, but pathogenic signatures are diluted systemically as a result of the strict compartmentalisation of immune responses. In this work, we aimed at defining immune mediators shared between the bloodstream and the synovial micro-environment, and relevant for disease activity in autoimmune arthritis.
Methods CD4+ T cells from blood and synovium of patients with juvenile idiopathic arthritis (JIA) were immunophenotyped by flow cytometry. The TCR repertoire of a circulating subset showing similarity with the synovium was analysed through next-generation sequencing of TCR β-chain CDR3 to confirm enrichment in synovial clonotypes. Finally, clinical relevance was established by monitoring the size of this subset in the blood of patients with JIA and rheumatoid arthritis (RA).
Results We identified a small subset of circulating CD4+ T cells replicating the phenotypical signature of lymphocytes infiltrating the inflamed synovium. These circulating pathogenic-like lymphocytes (CPLs) were enriched in synovial clonotypes and they exhibited strong production of pro-inflammatory cytokines. Importantly, CPLs were expanded in patients with JIA, who did not respond to therapy, and also correlated with disease activity in patients with RA.
Conclusions CPLs provide an accessible reservoir of pathogenic cells recirculating into the bloodstream and correlating with disease activity, to be exploited for diagnostic and research purposes.
- Autoimmunity
- Juvenile Idiopathic Arthritis
- Rheumatoid Arthritis
- Synovial fluid
- T Cells
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
The blood is the most easily accessible human tissue, and as such, it has been heavily investigated for pathogenic immune signatures in a variety of T cell-mediated diseases, including juvenile and adult forms of autoimmune arthritis. Notwithstanding its value in blood-borne diseases, such as HIV, investigations in the blood have not been equally fruitful for autoimmune rheumatological diseases. This is likely due to the strict compartmentalisation of tissue-restricted immune responses, which are shaped by the peripheral micro-environment and become highly diluted in the bloodstream.1–3 A good example of the immunological disconnect between the blood and the inflamed tissues is provided by past attempts at identifying pathogenic signatures in autoimmune arthritis by characterising the Vβ usage in synovial fluid versus blood.4 These studies were inconclusive because they were hindered by the limitations of the technology available at the time and by the inherent difficulties in matching the synovial repertoire with the much more diverse clonal representation of blood T cells.
To get insights in disease pathogenesis, the scientific community is moving away from immunological studies in the blood to embrace tissue immunology. Unfortunately, normal and inflamed human tissues are accessible only through invasive procedures.5 In addition, current therapies for autoimmune arthritis are often able to transiently reduce accumulation of synovial fluids at the affected joints, thereby reducing synovial sample availability and making analysis of the micro-environmental processes even more difficult.6 ,7
In this work, we first sought phenotypical patterns defining synovial T cells of patients affected by juvenile idiopathic arthritis (JIA) and adult rheumatoid arthritis (RA), and used this signature to select synovial-like T cells in the circulation. Then, we exploited next-generation sequencing to identify T cells based on their unique TCR, and used it as a barcode to demonstrate the identity between synovial T cells and their circulating counterparts. Finally, we proved that these circulating synovial-like T cells are associated with disease activity and responsiveness to therapy in human arthritis, expanding the clinical relevance of our discovery. Our work sheds new light on the relationship between micro-environmental and systemic immunity and might greatly facilitate the development of improved and targeted therapeutic approaches.
Methods
TCR repertoire
TCRβ CDR3 sequencing was performed by Adaptive Biotechnologies.8 ,9 Sorting was performed immediately after thawing. Dead cells were excluded using Live/Dead Fixable Near-IR Stain. Circulating pathogenic-like lymphocytes (CPLs) were sorted as human leucocyte antigen (HLA)-DR+CD14−CD3+CD4+CD25low/− cells from total peripheral blood mononucleated cells.
The analysis of TCR species composition is conceptually identical to the investigation of biodiversity. Since ecological methods are well developed, they have been properly adapted to TCR repertoire analyses.8–13 Each sample is interpreted as a distribution of individuals (T cell genomes) belonging to different species (TCR clonotypes), and can be interrogated for (i) its intrinsic diversity and (ii) its dissimilarity (ie, pairwise distance) from another sample. Diversity is determined by species richness (ie, the number of unique species relative to the population size) and distribution evenness: the higher the richness and the more evenly distributed the species, the higher the diversity. Likewise, the dissimilarity between samples is determined by the number of shared species, and their frequencies. We performed both types of analyses on productive nucleotide sequences (see online supplementary figure S4) and amino acid sequences derived by in silico translation (figure 3).
To properly address certain biological questions, cell subsets of varying size may be targeted for sampling.12 In our study, CPLs represent only a small fraction of blood CD4+ T cells; consequently, the number of T cell genomes obtained from CPL and CD4+ blood samples will differ of at least an order of magnitude. Taking into account that sample size is critical, because the higher the number of individuals sampled, the higher the probability of detecting rare species. In other words, the larger the sample size, the larger the diversity.12 This issue becomes apparent when two samples of different size are taken from the same population, the expectation being that they should display identical diversity, and no pairwise dissimilarity, except for the contribution of random sampling. By contrast, the larger sample will always show higher diversity than the smaller, and the two samples will appear more dissimilar than if they had the same size. This is because the larger samples will always detect rare species not comprised in the smaller sample, as a result of the imbalance in the number of examined individuals.12 Therefore, to ensure fairness of comparisons, it is necessary to contrast samples of equal size. To this aim, the smaller sample could be upsized to the size of the larger; alternatively, the larger could be downsized to the size of the smaller. The former strategy requires diversity and dissimilarity be extrapolated as a function of size, and is not used in practice. Conversely, the latter strategy is the standard in the field. To make assessments robust, sub-sampling is performed repeatedly, and the median of the readout of interest across all sub-samples is reported.10 ,12 ,13
For analyses of diversity, the Renyi entropy index was used.10 ,11 The larger the Renyi index, the larger the diversity. The Renyi index is modulated by the α parameter: a value of α above 1 puts more weight on abundant species, while a value below 1 puts more weight on rare species. Because, by definition, sampling of an entire population cannot be achieved, some TCR species will never be observed in the sample. Such under-sampling mostly affects rare TCRs. Therefore, the Renyi index must be evaluated across different values of α.10 ,11 Differential species diversity between two samples is substantiated only if the Renyi index is consistently greater (or lower) across all values of α. Equal sample size was achieved by repeated (200 times) sub-sampling, and the median of the Renyi index across sub-samples at each value of α was then plotted.10 ,12 ,13
For analyses of dissimilarity, two independent methods were applied. Using the first method, the pairwise distance between samples was calculated using the Chao-modified Jaccard index, which corrects for the effect of under-sampling.10 ,13 Using the second method, equal sample size was first achieved by repeated random sub-sampling.10 ,12 ,13 For each sample, we selected 200 sampling sizes at equally spaced intervals, corresponding to 200 increasing T cell genome counts, from a T cell genome count of 1 to the T cell genome count of the whole sample. At each sampling size, the selected number of individuals was drawn 200 times without replacement from the whole sample, and the fraction of unique synovial clonotypes shared with their paired blood samples, weighted for their frequencies, was calculated for each draw. The median percentage of overlap for each sampling size was then plotted, and the percentage of overlap was calculated at the size of the smaller sample to avoid extrapolation.10 ,12 ,13
The R code used for the analyses of the TCR repertoire is available as online supplementary information.
Results
Circulating CD4+ T cells from patients with JIA with active disease do not reflect the synovial pro-inflammatory signature
Our work was first kindled by the observation that circulating lymphocytes do not mirror their counterparts from the inflamed synovial micro-environment, which are enriched in pathogenic clonotypes.14–21 Figure 1A–D illustrates important discrepancies between blood and synovial T cells of patients with active JIA. Using flow cytometry, we inspected T cells for key processes involved in pathogenic T cell-mediated responses, that is, activation (CD69, CD25, HLA-DR), antigen recognition (Ki67, CD3), differentiation (CD45RA), migratory behaviour (CCR5, CCR6, CCR7) and maturation/exhaustion (CTLA-4, PD-1, LAG-3). Synovial T cells mainly comprised activated CD69+CD45RA− memory T cells, and were enriched in CD25+ and HLA-DR+ cells, while circulating T cells showed a mixed naive/memory resting phenotype (figure 1A, B). Consistent with recent antigen recognition, a large fraction of synovial T cells expressed Ki67 (an evidence of recent in vivo proliferation, thus a surrogate for antigen encounter) and down-regulated CD322 (figure 1B). Moreover, they preferentially expressed the pro-inflammatory chemokine receptors CCR5 and CCR6 over the lymphoid organ-homing CCR7 (figure 1C). Finally, synovial T cells exhibited signs of maturation/exhaustion, as revealed by the expression of CTLA-4, LAG-3 and PD-1, consistent with sustained antigen exposure (figure 1D).

Circulating CD4+ T cells from patients with active juvenile idiopathic arthritis (JIA) do not reflect the synovial pro-inflammatory signature. (A–D) Representative staining of synovial (black dotted lines) and peripheral blood (solid grey lines) samples of patients with active JIA, segregated by function. T cells were gated as CD14-CD3+CD4+CD25low/–FOXP3–. n=8. (E). Phenotypical results (MFI, median fluorescence intensity, for CD3, % positive for all other markers) were fed to an unsupervised hierarchical clustering algorithm and colour-coded upon row-wise normalisation. Each column represents an individual sample.
Altogether, these molecules compose a signature dramatically differentiating synovial and circulating T cells. Indeed, an unsupervised hierarchical clustering algorithm fed with this phenotypical data perfectly segregated the samples according to their tissue of origin (figure 1E).
From these data, it is evident that a much finer dissection is needed to distill complex pathogenic signatures from systemic compartments. However, the specific synovial T cell profile we identified might be used as a guide to identify pathogenic-like cells outside the inflamed synovium.
Circulating pathogenic-like lymphocytes (CPLs) from patients with active disease mirror the inflammatory synovial T cell signature
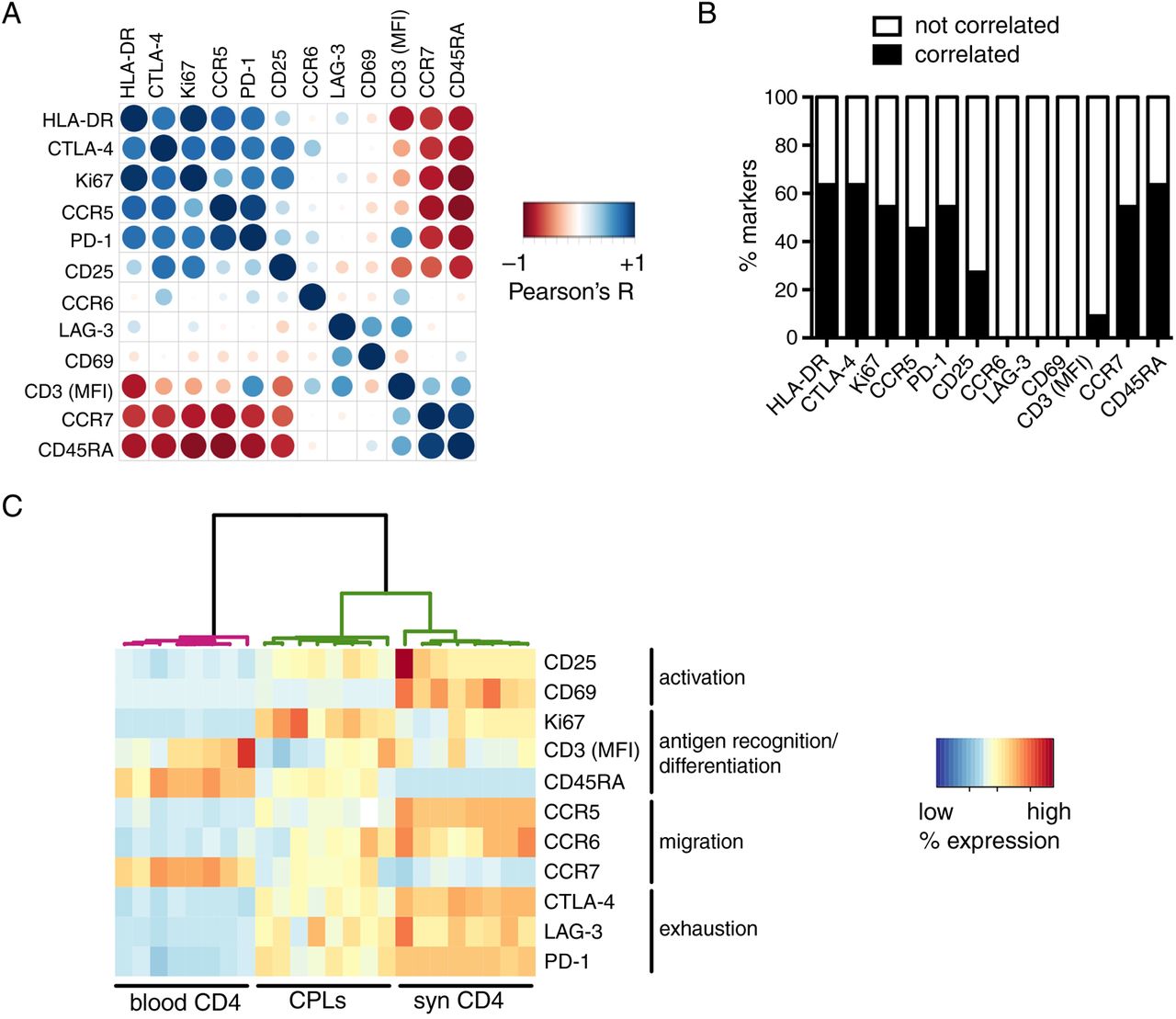
We hypothesised that if any fraction of pathogenic T cells recirculates out of the inflamed tissue, such population would have a phenotypical and functional profile similar to its synovial counterpart, enriched in arthritogenic T cells.14–21 Interestingly, a very small subset of blood T cells still expressed markers highly upregulated on synovial T cells (figure 1), indicating that the same signature is detectable also in the blood, only much more diluted than in the synovium. In the blood, we observed high pairwise correlations between most of the markers (figure 2A, B). Some notable exceptions were CD69 and LAG-3, whose expression did not correlate with that of any other molecule, and CD25, which correlated with some but not all. By contrast, another activation marker, HLA-DR, was one of the most correlated (figure 2A, B). These findings are in line with the differential expression kinetics of activation markers; indeed, its late upregulation when compared with CD6923 ,24 enables HLA-DR to still remain detectable when activated T cells reach the blood. These data indicate that different T cell activation markers have unique features; as such, they cannot be interchangeably used as proxies for peripheral inflammation.

Circulating pathogenic-like lymphocytes (CPLs) from patients with active disease mirror the inflammatory synovial T cell signature. (A) Pairwise correlations (Pearson's R) were calculated for the indicated markers (median fluorescence intensity (MFI) for CD3, % positive for all other markers) measured on circulating CD4+ T cells of patients with active juvenile idiopathic arthritis (JIA), and then colour-coded as indicated in the legend and re-ordered according to the aggregate level of correlation (first principal component). Larger circles indicate higher |R|. n=32. (B) For each marker from panel A, the percentage of pairwise comparisons with all other markers from the panel ensuing in correlation (|R| ≥0.7), or no correlation (|R|<0.7), is reported. (C) Phenotypical results (MFI for CD3, % positive for all other markers) within gated CPLs and the bulk of blood or synovial CD4+ T cells from patients with JIA with active disease were fed to a hierarchical clustering algorithm and colour-coded upon row-wise normalisation. A white cell indicates data not available. n=8 per group.
An important practical consequence of the high intercorrelations between most markers composing the signature of synovial-like T cells is that any of these proteins might be used to pinpoint this population. This provides the opportunity to isolate it with a single marker, thus reducing technical complexity in view of the potential clinical application of our approach. Among the molecules tested, HLA-DR was particularly appealing as a sorting marker, because (1) it is a surface protein, thereby allowing the isolation of living cells and downstream functional analyses, in contrast to other relevant intracellular antigens that require fixation, such as Ki67 and CTLA-4; (2) differently from other proteins, including PD-1 and chemokine receptors, it is seemingly devoid of signalling activity in T cells, enabling the sorting of unperturbed T cells. Several studies reported conflicting results on HLA-DR signalling capabilities.25–32 In our hands, cross-linking of HLA-DR in T cell lines did not elicit calcium flux (see online supplementary figure S3A), a key signalling event downstream of HLA-DR triggering in dendritic cells.32 In addition, HLA-DR+ T cell lines did not induce proliferation of allogeneic CD4+ T cells (see online supplementary figure S3B). Altogether, these data demonstrate that HLA-DR does not exhibit any back-signalling activity and does not endow T cells with antigen-presenting capabilities. Therefore, HLA-DR can be used to sort viable and unperturbed T cells.
We sought to confirm that HLA-DR could indeed be used to identify circulating T cells similar to synovial T cells. Differently from the bulk of blood CD4+ T cells, HLA-DR-gated T cells were enriched in activated, memory-like, antigen-experienced Ki67+ cells; in addition, they might have been exposed to autoantigens in affected joints, as they preferentially expressed CCR5 and CCR6 over CCR7, and they also expressed high levels of maturation/exhaustion markers (figure 2C). Importantly, when these phenotypical data were analysed through unsupervised clustering, the algorithm partitioned HLA-DR-gated cells apart from blood T cells, but together with synovial T cells (figure 2C).
Altogether, our data demonstrate that HLA-DR can be used to identify a small population of ‘circulating pathogenic-like lymphocytes’ (CPLs) expressing the typical synovial T cell signature. CPLs comprise activated, antigen-experienced T cells able to recirculate through inflamed sites.
CPLs are enriched in clonotypes of synovial T cells
To get a compelling proof of the parallel between CPLs and synovial T cells, we compared their TCR repertoires through next-generation sequencing of TCRβ CDR3, the main determinant of TCR specificity. This approach enabled us to verify T cell identity at clonal level (ie, at the level of a single CDR3 sequence). We were able to sequence an average number of 71 500 blood T cells, 5000 CPLs and 15 000 synovial T cells.
First, we evaluated the TCR repertoire diversity by calculating the Renyi index.10 ,11 In accordance with previous reports of synovial T cell oligoclonality,33 the TCR diversity in the synovium was lower than in the blood. Importantly, also CPLs were less diverse compared with the bulk of blood T cells—and in some instances as oligoclonal or even more oligoclonal than the synovium—at the level of both amino acid and nucleotide sequences (figure 3A, B and see online supplementary figure S4A,B, respectively). These results demonstrate that CPLs display reduced TCR diversity, similarly to synovial T cells.33

Circulating pathogenic-like lymphocytes (CPLs) are enriched in clonotypes of synovial T cells. Next-generation sequencing of TCRβ CDR3 sequences was performed on blood or synovial CD4+ T cells of patients with active juvenile idiopathic arthritis (JIA). All data refer to amino acid sequences derived by in silico translation. (A and B) Renyi diversity indices for different values of α (A) and summary at α=1 (B; grey dashed line in panel A). Differential species diversity is substantiated only if the Renyi index is consistently greater (or lower) across all values of the α parameter. (C) Clustering based on TCR repertoire distances, calculated as 1—Chao-modified Jaccard index (Chao distance). (D) Summary of the TCR repertoire pairwise Chao distances between CPL or blood samples and their paired synovial sample. (E) Overlap of the TCR repertoire from CPL and blood samples with their matched synovial sample. Each open circle is the median overlap of 200 subsamples of the indicated size from either CPL or blood samples with 200 subsamples of the same size from their paired synovial sample. (F) Summary statistics for the overlap data, as measured at the size of the smaller sample (grey dashed line in panel E). In A, C and E, each panel represents an individual patient.
Next, we investigated how close the TCR repertoire of CPLs and synovial T cells are at clonal level. Upon clustering based on the dissimilarity of distribution of the TCR sequences,13 CPLs segregated together with synovial T cells and apart from blood T cells (figure 3C, D and see online supplementary figure S4C, D). Accordingly, CPLs shared a substantially higher fraction of CDR3 sequences with synovial TCRs than blood T cells (figure 3E, F and see online supplementary figure S4E, F, respectively). Importantly, the conspicuous difference observed between CPLs and the total peripheral blood T cells was completely overlooked in a traditional analysis of Vβ family usage (see online supplementary figure S4G), which lacks the resolution necessary to finely dissect the clonal composition of the TCR repertoire.
In summary, these data demonstrate that CPLs comprise a large fraction of synovial clonotypes.
CPLs retain their pro-inflammatory potential
As CPLs are highly enriched in T cells recirculating through the inflamed synovial micro-environment, which is, in turn, enriched in arthritogenic T cells,14–21 we investigated whether they would retain their pro-inflammatory characteristics while recirculating in peripheral blood. Importantly, CPLs produced much higher levels of the pro-inflammatory cytokines interferon-γ, interleukin-17 and tumour necrosis factor-α than the bulk of blood CD4+ T cells (figure 4), demonstrating that they are able to actively fuel inflammation.

CPLs retain their pro-inflammatory potential. Cytokine production from circulating CD4+ T cells of patients with active JIA upon PMA/ionomycin stimulation. Each line corresponds to an individual patient. CPLs, circulating pathogenic-like lymphocytes; JIA, juvenile idiopathic arthritis; IFN, interferon; IL, interleukin; PMA, phorbol 12-myristate 13-acetate; TNF, tumour necrosis factor.
CPLs correlate with unresponsiveness to therapy and disease activity in both juvenile and adult autoimmune arthritis
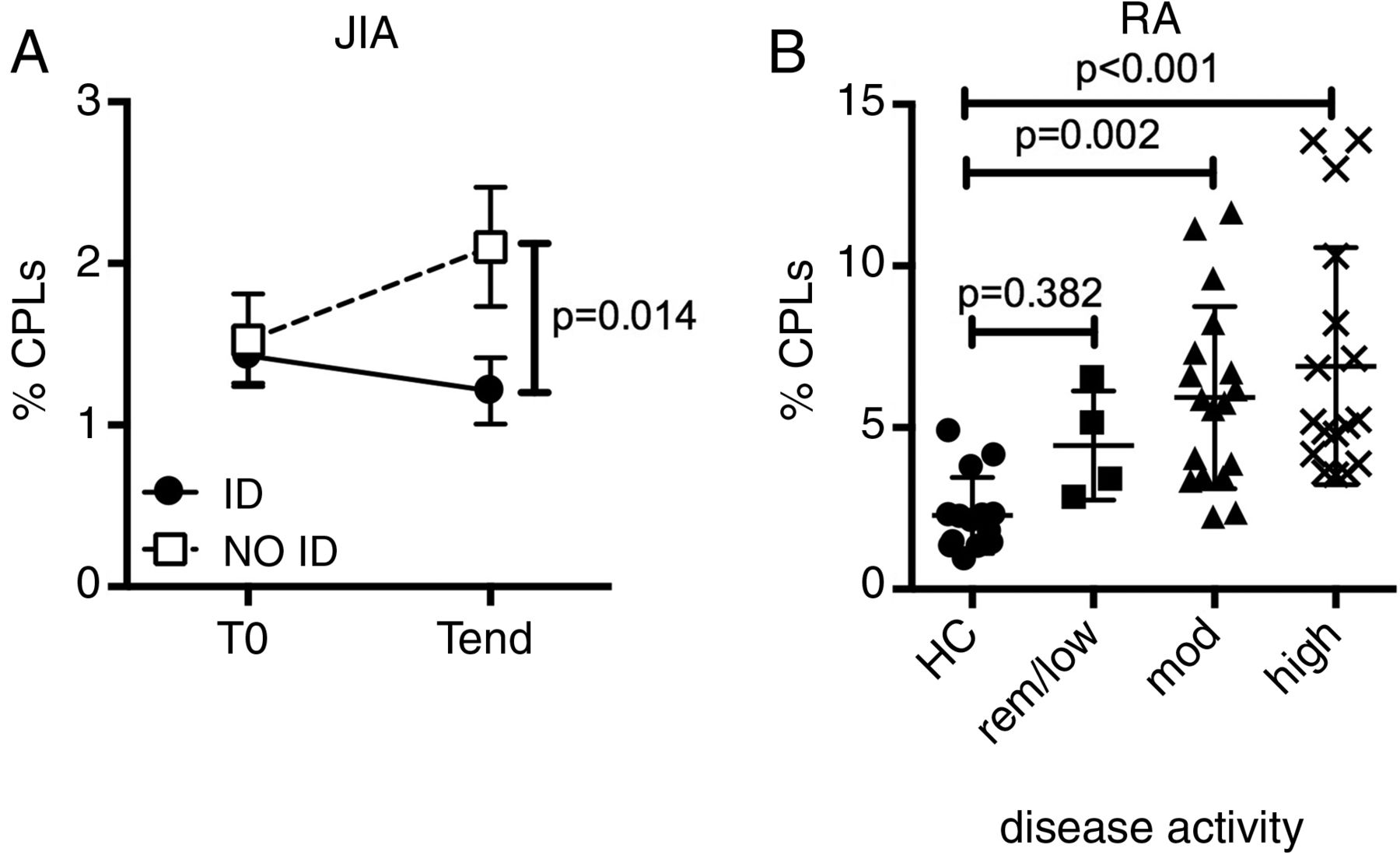
Our discovery of a circulating pool of pathogenic-like T cells might have profound clinical relevance. Indeed, it might provide the missing link between the clinical manifestations of the disease in the periphery and the seemingly normal systemic immunity. Under this framework, the frequency of CPLs in the periphery should correlate with disease activity. We tested this hypothesis on peripheral blood samples from patients with JIA, collected before (T0) and after (Tend) therapy,34 and stratified for responsiveness to therapy based on whether they reached inactive disease (ID)35 or not (NO ID) at Tend. CPLs slightly declined in prospective ID patients, but substantially increased in NO ID patients, resulting in NO ID patients harbouring a doubled frequency of CPLs at Tend when compared with ID patients (figure 5A). Thus, the increase in CPL frequency mirrors the persistent or intensified synovial inflammation clinically observed in NO ID patients.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Circulating pathogenic-like lymphocytes (CPLs) correlate with unresponsiveness to therapy and disease activity in both juvenile and adult autoimmune arthritis. (A) Percentage of CPLs over time in blood CD4+ T cells from patients with juvenile idiopathic arthritis (JIA). All patients were in active disease at T0 and then segregated based on their clinical activity at Tend. ID=inactive disease; NO ID: not inactive disease. Vertical lines represent SEM. n=10–15 per group, per time point. (B) CPL representation in blood CD4+ T cells from patients with rheumatoid arthritis (RA), segregated by disease activity score (DAS-28 3) and compared with healthy controls (HC). low/rem: low/remission (DAS≤3.2); mod: moderate (3.2<DAS≤5.1); high: DAS>5.1. Each dot represents an individual patient.
We then asked whether this finding, unequivocal in JIA, might be extended to other autoimmune diseases. Therefore, we examined adult RA36 for the presence of CPLs. Importantly, CPLs were expanded in patients with RA when compared with healthy controls, and substantially increased with worsening activity score (figure 5B). This observation confers a broader clinical significance to CPLs beyond paediatric autoimmunity.
Discussion
In this work, we identified a small population of pathogenic-like T cells that is (1) reproducing the synovial T cell signature in the circulation; (2) able to recirculate through the site of autoimmune reaction; (3) highly enriched in synovial clonotypes; and (4) correlating with disease activity in both juvenile and adult autoimmune arthritis. Our discovery exposes an easily accessible pool of pathogenic-like cells, which may be used to substantially advance our understanding of aberrant self-recognition, as well as to design targeted diagnostic and therapeutic tools.
Our claims are supported by the functional profile of CPLs, and then formally substantiated by their TCR repertoire. Indeed, CPLs recapitulate the synovial T cell signature, being enriched in proliferating Ki67+ cells and preferentially expressing pro-inflammatory chemokine receptors and exhaustion markers, in the absence of overt infection. In addition, CPLs display lower levels of CD3 compared with the bulk of blood T cells, a sign of TCR-mediated activation, as opposed to cytokine-mediated bystander activation.22 The analysis of their TCR repertoire provides direct evidence in support of this claim: indeed, CPLs are oligoclonal and share a consistent fraction of clonotypes with the synovium, where arthritogenic T cells reside.14–21 Importantly, the overlap increases as a function of the number of sequenced T cells. Therefore, at the level of the entire CPL repertoire, the percentage of clonotypes shared with the synovium is even higher than the one we measured on a sample (5000 CPLs on average from less than 10 mL of blood). Overall, these findings indicate that CPLs include T cells identical to those residing in the inflamed micro-environment, and still retain their pro-inflammatory features while recirculating.
CPL features are consistent with the hypothesis that they escape from the site of autoimmune reaction. Alternatively, it is possible that CPLs recirculate in the blood after being primed/reactivated in the lymph nodes draining auto-reactive sites, and are thus migrating to, rather than from, the inflamed joints. There is a third scenario, which would reconcile both alternatives. Chronic autoimmunity is often accompanied by the appearance of tertiary lymphoid structures within autoreactive sites.37 These hybrid structures show elements of both peripheral tissues and lymph nodes, and support direct antigen presentation and T cell activation. In vivo cell tracking would be needed to distinguish between these possibilities. Unfortunately, class II MHCs are only expressed by antigen-presenting cells but not by T cells in mice, due to a mutation in the class II transactivator gene.38 This limitation underscores the need for exquisitely human studies, such as our own, to avoid overlooking important aspects of disease pathogenesis due to a disconnect between the human disease and animal models.
Although CPLs are present in paediatric ID patients and adult healthy individuals, consistent with a basal level of possibly self-reactive clonotypes even in the absence of disease,39 they are selectively expanded in NO ID patients and patients with active RA. Thus, CPLs provide a correlate and suggest a potential interpretation for unresponsiveness to therapy and disease activity for future mechanistic studies. Indeed, the increase in CPL frequency may result from the synovial expansion of arthritogenic T cells resistant to suppression and therapy, which boost synovial inflammation, ultimately fostering the clinical manifestations of the disease. Given their ability to actively produce inflammatory cytokines, CPLs might also contribute directly to the spreading of the disease to unaffected joints and to the exacerbation of systemic symptoms.
CPLs are defined by their synovial-like T cell signature, but they can be isolated with a single marker, substantially simplifying their investigation and isolation in clinical settings. Our strategy for the identification of arthritogenic T cells bypasses the limitations of current approaches based on tetramers or restimulation, namely: (1) the need for prior knowledge of antigen specificity, a major hurdle in autoimmune diseases such as JIA, for which most auto-antigens have yet to be identified;5 ,40 and (2) the ability to detect only TCR clones with high affinity for any given autoantigen.41 In addition, the small amount of blood withdrawn from children affected by JIA imposes serious additional constraints on the number of antigens that can be tested on a single sample. These issues motivated our search for alternative and more effective strategies to identify pathogenic T cells in the circulation. We first moved from the consideration that synovial T cells are enriched in arthritogenic T cells.14–21 Thus, we took advantage of recent developments in TCR repertoire profiling to identify synovial clonotypes in the bloodstream. Differently from traditional methods, the next-generation sequencing of TCR CDR3 regions provides the opportunity to perform investigations at clonal level, enabling us to establish the genetic overlap between CPLs and synovial T cells.
Differently from blood-borne diseases, where it is easy to envision that pathogenic T cells can be easily isolated from the blood, uncovering a small but detectable pool of circulating pathogenic-like T cells in an autoimmune disease was unexpected. Given the small size of this pool, it is easy to appreciate why the search of arthritogenic signatures in whole blood has been disappointing. Indeed, the majority of T cells (∼90%–98%) display negligible similarity to their synovial counterparts, in contrast to the tiny fraction (∼2%–10%) of CPLs, which also correlate with disease activity. Therefore, when blood is investigated as a whole, most molecular events underlying the mechanisms of disease will be too diluted to emerge from noise. Our study provides researchers with the right target population, thereby removing from measurements that irrelevant background ultimately responsible for holding back progress in the understanding of autoimmune arthritis.
We first discovered CPLs in JIA, and then confirmed their presence and clinical relevance also in RA. An important direction for future studies is investigating the role of CPLs in other autoimmune diseases. These studies may prove particularly useful for conditions with difficult or no access to the site of inflammation, such as multiple sclerosis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online tcr-analysis
- Data supplement 4 - Online tcr-primary-data
Footnotes
Handling editor Tore K Kvien
RS and MR contributed equally.
Contributors RS and MR equally contributed to study design, experiments, data collection, analysis and interpretation, statistical analyses and writing of the manuscript. JvL contributed to some experiments and writing of the manuscript. CAW, MM, SM-M, MG, AM and DJL contributed to sample collection, data interpretation and writing of the manuscript. SA contributed to study design and oversight, data interpretation and writing of the manuscript.
Funding Work was supported by NIAMS (1R01 AR049762), the Bartman Foundation, the NMRC (NMRC/STaR/020/2013; Ministry of Health, Singapore) and Singhealth. JvL was supported by the Dutch Arthritis Foundation.
Competing interests None.
Ethics approval IRB of the Sanford-Burnham Medical Research Institute.
Provenance and peer review Not commissioned; externally peer reviewed.