Article Text

Abstract
Objectives To explore the functional basis for the association between ankylosing spondylitis (AS) and single-nucleotide polymorphisms (SNPs) in the IL23R-IL12RB2 intergenic region.
Methods We performed conditional analysis on genetic association data and used epigenetic data on chromatin remodelling and transcription factor (TF) binding to identify the primary AS-associated IL23R-IL12RB2 intergenic SNP. Functional effects were tested in luciferase reporter assays in HEK293T cells and allele-specific TF binding was investigated by electrophoretic mobility gel shift assays. IL23R and IL12RB2 mRNA levels in CD4+ T cells were compared between cases homozygous for the AS-risk ‘A’ allele and the protective ‘G’ allele. The proportions of interleukin (IL)-17A+ and interferon (IFN)-γ+ CD4+ T-cells were measured by fluorescence-activated cell sorting and compared between these AS-risk and protective genotypes.
Results Conditional analysis identified rs11209032 as the probable causal SNP within a 1.14 kb putative enhancer between IL23R and IL12RB2. Reduced luciferase activity was seen for the risk allele (p<0.001) and reduced H3K4me1 methylation observed in CD4+ T-cells from ‘A/A’ homozygotes (p=0.02). The binding of nuclear extract to the risk allele was decreased ∼3.5-fold compared with the protective allele (p<0.001). The proportion of IFN-γ+ CD4+ T-cells was increased in ‘A/A’ homozygotes (p=0.004), but neither IL23R nor IL12RB2 mRNA was affected.
Conclusions The rs11209032 SNP downstream of IL23R forms part of an enhancer, allelic variation of which may influence Th1-cell numbers. Homozygosity for the risk ‘A’ allele is associated with more IFN-γ-secreting (Th1) cells. Further work is necessary to explain the mechanisms for these important observations.
- Ankylosing Spondylitis
- Gene Polymorphism
- T Cells
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Ankylosing spondylitis (AS) is the prototypic spondyloarthropathy (SpA), characterised by prominent axial skeletal involvement and enthesitis.1 Genome-wide association studies (GWAS) have clearly demonstrated the polygenic nature of AS.2–4 Further, many of the genes that have been implicated are also associated with conditions like inflammatory bowel disease (IBD) and psoriasis that occur much more commonly in individuals with AS than the general population.5 ,6 Of these, IL23R (encoding the specific portion of the heterodimeric interleukin (IL)-23 receptor) was the first to be associated with AS.7 More than 40 loci have now been implicated in AS, several of which (eg, IL23R, IL12B, IL6R, TYK2, IL27R, IL1R2, IL1R1 and STAT3) potentially affect IL-23-driven pro-inflammatory pathways.2–4 ,8 ,9 The importance of these pathways is further highlighted by the potential role of IL23R-expressing cells at the entheses in murine models of SpA.10 The primary IL23R association with AS (also psoriasis and IBD) is with rs11209026, a missense variant (Arg381Gln) in the cytoplasmic tail, which alters IL-23R signalling.7 ,11 ,12 In addition, a second independent association signal has been identified in the intergenic region downstream of IL23R and upstream of IL12RB2 (encoding the 130kD β2 chain specific to the IL-12 receptor).3 This second signal is also associated with IBD.5 Currently, the mechanism underlying the latter association is unknown. In this study, we have identified a putative regulatory element (PRE) between IL23R and IL12RB2. We have then investigated the impact of AS-associated single-nucleotide polymorphisms (SNPs) on the function of this PRE.
Methods
Identification of a PRE
We used a combination of published AS GWAS data3 and epigenetic data from the ENCODE13 and Roadmap Epigenomics Projects,14 to identify a 1.14 kb PRE between IL23R and IL12RB2, including the AS-associated SNPs rs11209032 and rs6677188. The epigenetic data included DNase I hypersensitivity sites, transcription factor (TF) binding sites and histone modifications.
Patients with AS
All patients in these studies fulfilled the modified New York AS criteria15 or ASAS axial SpA imaging criteria.16 Following informed consent, blood samples for the functional studies (below) were obtained from patients.
IFN-γ+ and IL-17A+ T-cell FACS analysis
Blood samples were obtained from 52 biologic-naive AS cases (mean age 42 years±SD 12.3). The mean Bath AS disease activity index (BASDAI) was 4.6±SD 2.2.
Gene expression
Blood samples were obtained from 12 AS cases (mean age 61.5 years±SD 12.6). The mean BASDAI was 3.4 (±SD 1.7) and mean C reactive protein 7.1 mg/L (±SD 6.4). Only nine were currently taking non-steroidal anti-inflammatory analgesics, and none were taking corticosteroids or other immunomodulatory drugs.
Genotyping
Historical typing data from previously published AS Immunochip study3 were used if available or were obtained using TaqMan Genotyping Assay (Life Technologies, Paisley, UK) to assign SNP genotypes. Where required, DNA was extracted from peripheral blood mononuclear cells (PBMCs) using the QIAGEN AllPrep DNA/RNA Mini Kit (QIAGEN).
CD4+ T-cell isolation
CD4+ T-cells were isolated from PBMCs using the negative selection CD4+ T-cell Isolation kit (Miltenyi, Bisley, Surrey, UK). CD4+ T-cells were plated for 4 h/overnight in Roswell Park Memorial Institute supplemented with 10% fetal bovine serum before harvesting for experiments. Cell viability was checked with trypan blue or fluorescence-activated cell sorting (FACS) analysis.
FACS analysis
PBMCs were isolated by density gradient centrifugation using Histopaque (Sigma, Dorset, UK), frozen and stored in liquid nitrogen before staining. Intracellular cytokine staining of Th17 and Th1-cells was carried out using BD Cytofix/Cytoperm kit (BD Bioscience, Oxford, UK). Cells were stimulated with 100 ng/mL phorbol 12-myristate 13-acetate (PMA) (Sigma, Dorset, UK) and 1 μg/mL ionomycin (Sigma, Dorset, UK) for 4 h in the presence of Golgi STOP and Golgi plug. After surface staining using CD3-BV605, CD4-APC and CD8-BV510 antibodies (Biolegend, London, UK), cells were fixed and permeabilised, then stained with IL-17A-FITC (eBiosciences, Ireland, UK) and interferon (IFN)-γ-AF700 (Biolegend). Dead cells were excluded using Fixable Viability Dye eFluor 780 (eBiosciences). Representative FACS plots of the gating strategy and intracellular staining are shown in online supplementary figure S1.
Supplementary figures
Electrophoretic mobility shift assay
Nuclear extract from HEK293 cells (human embryonic kidney cell line) was purchased from Active Motif (Belgium, Germany). Electrophoretic mobility shift assays (EMSAs) were performed with LightShift Chemiluminescent EMSA Kit (Thermo Scientific, Waltham, USA) using 5 μg of nuclear extract, and 10 fmol biotin labelled double-stranded oligonucleotides (50 bp fragment—Eurofins, Wolverhampton UK). The sequences of the synthetic single-stranded oligonucleotides used in the construction of these double-stranded oligonucleotides are listed in the online supplementary methods. Single-stranded biotinylated oligonucleotides were mixed and annealed at room temperature for 1 h. Unlabelled competitor probes were in 100-fold excess. EMSAs were performed according to standard protocol (Thermo Scientific). The involvement of TWIST1 in these DNA-protein complexes was investigated by including TWIST1 antibody (ab50877—Abcam, Cambridge, UK). The results were confirmed in five independent experiments. Band intensity from these five experiments was measured by ImageJ Launcher (V.1.4.3.67). A detailed protocol is available in online supplementary methods.
Supplemental material
Luciferase reporter assay
The 1.14 kb PRE sequence was amplified from genomic DNA and cloned into TA cloning kit pCR2.1 vector (Invitrogen, Paisley, UK). It was subcloned into pGL4.23 [luc2/minP] reporter vector (Promega, Madison, USA) at the SacI/XhoI restriction sites upstream of the minimal promoter necessary to drive the luciferase reporter gene (primer sequences available on request). Point mutations corresponding to genetic variants (G/A) of rs11209032 were introduced using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent, Santa Clara, USA). Luciferase reporter assay details are available in online supplementary methods.
Quantitative real-time RT-PCR
RNA was isolated from CD4+ T-cells from patients of different genotypes (PMA and ionomycin stimulated; overnight). RNA isolation was performed using the Allprep DNA/RNA Mini kit (QIAGEN) and cDNA synthesis (for 500 ng RNA) was prepared with Superscript III from Invitrogen. A final concentration of 5 ng was used in quantitative PCR (qPCR), which was performed with the ABI ViiA7 PCR instrument (Applied Biosystems, Paisley, UK) using SYBR Master mix (Applied Biosystems) with evaluation of dissociation curves. mRNA levels of each gene were quantified using the DDCt method and expressed relative to β-actin. For each gene, a TaqMan Gene Expression Assay was used (Life Technologies—according to the manufacturer's instructions); IL23R (Hs00332759_m1), IL12RB2 (Hs00155486_m1), IFNG (Hs00989291_m1), IL10 (Hs00961622_m1) and β-actin (Hs01060665_g1). H19 primers were purchased from Diagenode (Liege, Belgium).
Statistical analysis
Association data for genotyped SNPs were obtained on the subset of AS cases of white British ancestry and white British controls from Immunochip GWAS.3 To evaluate the presence of independent effects on genetic susceptibility at the IL23R-IL12RB2 intergenic region, we performed conditional analysis on 4230 AS cases and 9700 matched controls, as previously described.3 Association analysis was performed using the logistic regression function in PLINK, V.1.90, accounting for population structure with 10 principal components.17 One-way analysis of variance and two-tailed Student's t test were used to determine statistical significance using GraphPad Prism (V.5.03).
Results
Identification of a PRE within IL23R-IL12RB2 intergenic region associated with AS
We identified a PRE (Chr1:67739940–67741075) located 14.2 kb downstream of IL23R and 33.1 kb upstream of IL12RB2 (figure 1). This PRE overlaps a region reported in the Immunochip study to be independently associated with AS after conditioning on association at the primary rs11209026 coding SNP.3 This region is DNase I hypersensitive (in Th1-cells, but not Th17-cells) and exhibits TF binding (lymphoblastoid cell line; GM12878) and enhancer-associated H3K4me1 methylation (CD4+ CD25− IL-17A− T-cells; PMA and ionomycin-stimulated).1 ,3 ,14
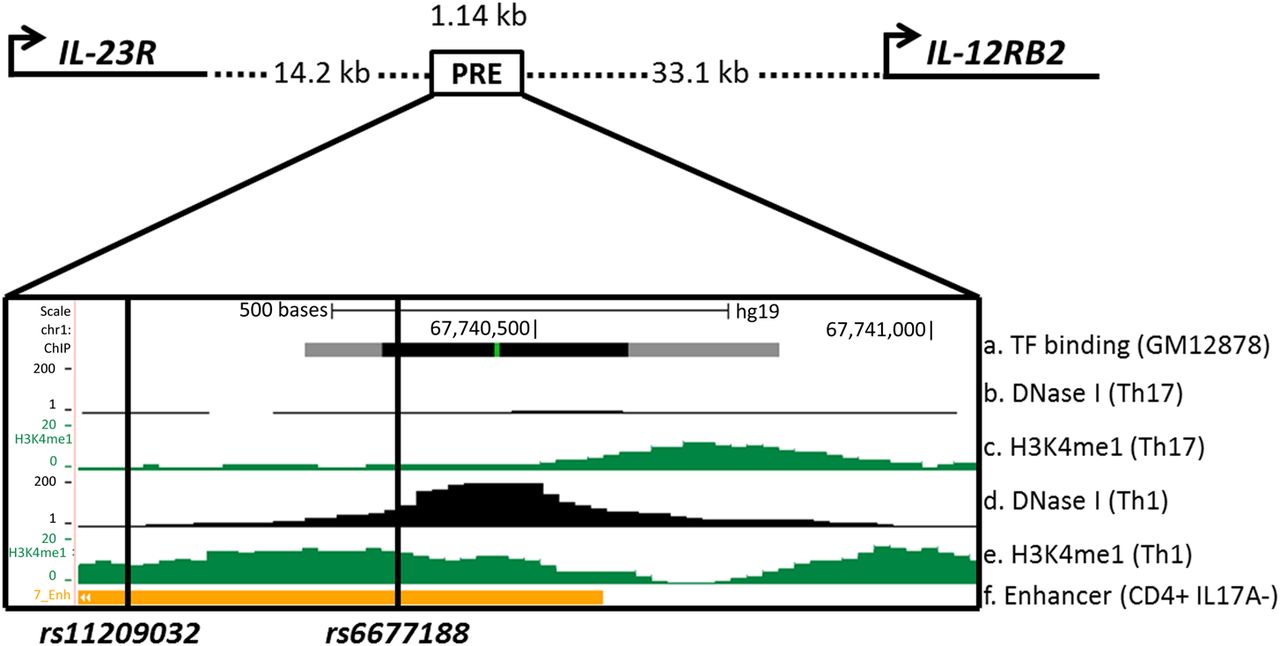
Epigenetic and transcriptional landscape of the 1.14 kb putative regulatory element (PRE) containing rs11209032 and rs6677188 downstream of IL23R. Cartoon representation of IL23R and IL12RB2 promoter and PRE location (Chr1:67739940–67741075). ENCODE and Roadmap data: (A) Transcription factor (TF) binding sites (grey/black box) in lymphoblastoid cell line GM12878. (B) DNase I hypersensitivity in Th17-cells. (C) H3K4me1 methylation in Th17-cells. (D) DNase I hypersensitivity in Th1-cells. (E) H3K4me1 methylation in Th1-cells. (F) Enhancer chromatin state in CD4+ CD25− interleukin (IL)-17A− T-cells (PMA and ionomycin-stimulated).
Conditional analysis identifies rs11209032 as a candidate casual variant
The PRE contains 10 SNPs, 3 of which show strong or weak association with AS; rs11209032 (p=5.8×10−16), rs80216366 (p=4.9×10−21) and rs6677188 (p=2×10−4). Conditional analysis previously performed on the primary IL23R coding SNP rs11209026 revealed no residual association at rs80216366 (p=0.4), indicating that this association is due to linkage disequilibrium (LD) with rs11209026.3 The associations at rs11209032 (p=1.5×10−9) and rs6677188 (p=9.3×10−7) were robust to conditioning on rs11209026, revealing a second region of independent association.3 We then performed further conditional analysis on rs11209032 or rs6677188, respectively, and found that the association at rs6677188 disappeared after conditioning on rs11209032, indicating that this association was due to LD with rs11209032 (table 1). In contrast, the strong association with rs11209032 was retained (p=8.1×10−12) after conditioning on rs6677188, thereby establishing the primacy of the rs11209032 association with AS in this region. However, a statistical association does not necessarily guarantee functionality. Initial interrogation of publicly available datasets suggested that rs6677188 might actually be more functionally relevant than rs11209032 (despite the genetic association data above). rs6677188 overlaps DNase I hypersensitivity (in Th1-cells) and TF binding sites (in GM12878 cells), whereas rs11209032 lies on the border of these sites (see figure 1). In the following functional study, we therefore analysed both SNPs.
Conditional analysis of single-nucleotide polymorphism (SNP) associations at IL23R-IL12RB2 intergenic region
Homozygosity for the AS-risk allele is associated with increased Th1-cell frequencies
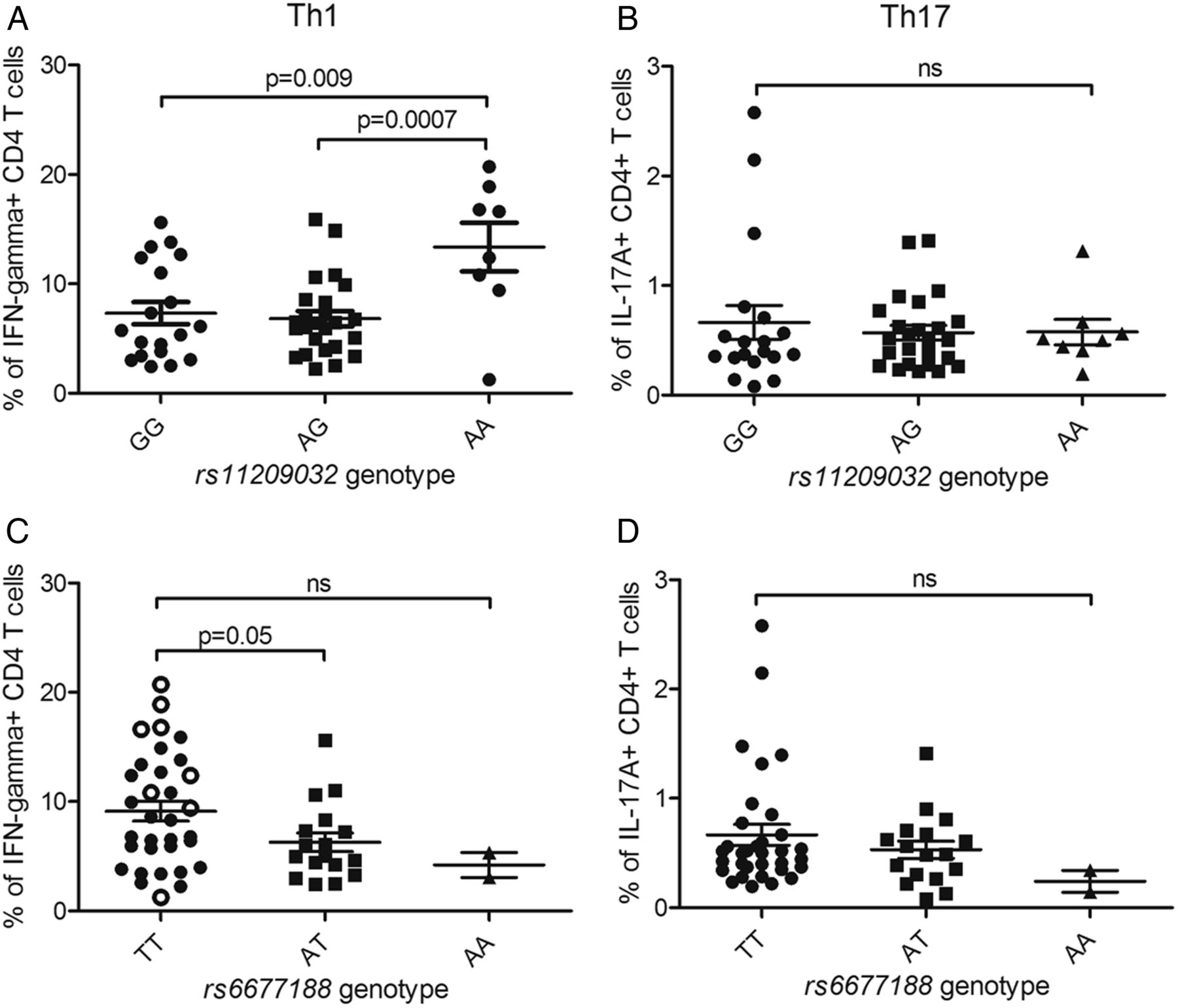
We sought to determine the mechanisms by which rs11209032 or rs6677188 affect AS susceptibility. The PRE containing rs11209032 and rs6677188 lies between IL23R (expressed on Th17-cells) and IL12RB2 (expressed on Th1-cells). Therefore, we measured the frequencies of Th17- and Th1-cells from patients with known SNP genotypes. Th17-cell and Th1-cell frequencies were measured by FACS for IL-17A+ and IFN-γ+ CD4+ T-cells, respectively (figure 2). A significant increase in the percentage of IFN-γ+ CD4+ T-cells was observed in patients homozygous for the ‘A’ (risk) allele at rs11209032 (figure 2A, p<0.01), but not for IL-17A+ CD4+ T-cells (figure 2B). The ‘T/T’ genotype at rs6677188 is weakly associated with an increased percentage of IFN-γ+ CD4+ T-cells (p=0.05, figure 2C), but not IL-17A+ CD4+ T-cells (figure 2D). However, this observed increase in the percentage of IFN-γ+ CD4+ T-cells apparently associated with rs6677188 is actually secondary to the association with the rs11209032 ‘A’ (risk) allele, which is in complete LD with the rs6677188 ‘T’ allele. If the rs11209032 ‘A/A’ homozygotes are excluded from the rs6677188 analysis (open circles in figure 2C), the apparent difference disappears. There was no correlation between the frequency of double-positive IL-17A+/IFN-γ+ CD4+ T-cells and the SNP genotype (see online supplementary figure S2). Overall, these results are consistent with the conditional analysis in suggesting that rs11209032 is the most probable causal variant (through effects on Th1-cell differentiation). Support for an effect in Th1-cells comes from publicly available data sets that show the presence of DNase I hypersensitivity in Th1-cells within the PRE, but not in Th17-cells (see figure 1). Consequently, all the subsequent experiments focused on rs11209032 alone.

Homozygosity for the ankylosing spondylitis (AS)-risk allele at rs11209032 is associated with increased Th1-cell frequencies. (A) The % of interferon (IFN)-γ+ CD4+ T-cells in patients with AS of each genotype at rs11209032 (19 GG, 25 GA, 8 AA). (B) The % of interleukin (IL)-17A+ CD4+ T-cells in patients with AS of each genotype at rs11209032 (19 GG, 25 GA, 8 AA). (C) The % of IFN-γ+ CD4+ T-cells in patients with AS of each genotype at rs6677188 (33 TT, 17 TA, 2 AA). Patients ‘A/A’ at rs11209032 are highlighted by open circles. (D) The % of IL-17A+ CD4+ T-cells in patients with AS of each genotype at rs6677188 (33 TT, 17 TA, 2 AA). The percentage of cells is expressed as mean±SEM. Student's t test was used.
Differential binding of nuclear extract at rs11209032
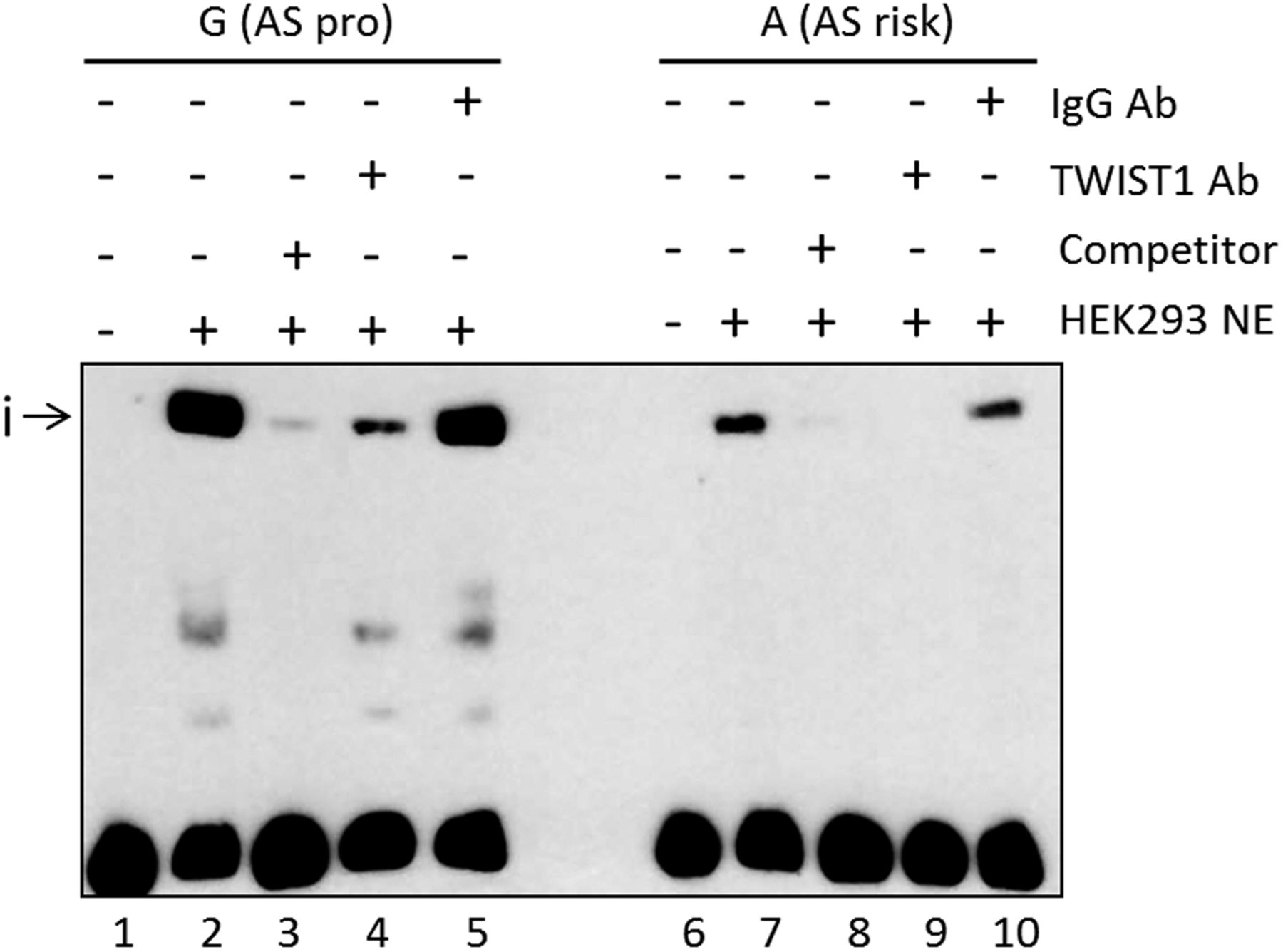
We investigated the mechanism for the increase in IFN-γ+ CD4+ T-cells (above) by looking for effects of rs11209032 on TF binding. EMSAs were performed using nuclear extract from HEK293 cells (human embryonic kidney cell line). The addition of nuclear extract to a 50mer DNA probe containing rs11209032 revealed a DNA-protein complex (i), with ∼3.5-fold lower binding to the ‘A’ (risk) allele (figure 3, p<0.001). Binding to the ‘G’ (protective) allele was outcompeted by 100-fold excess of unlabelled ‘G’ (figure 3 and online supplementary figure S3A).
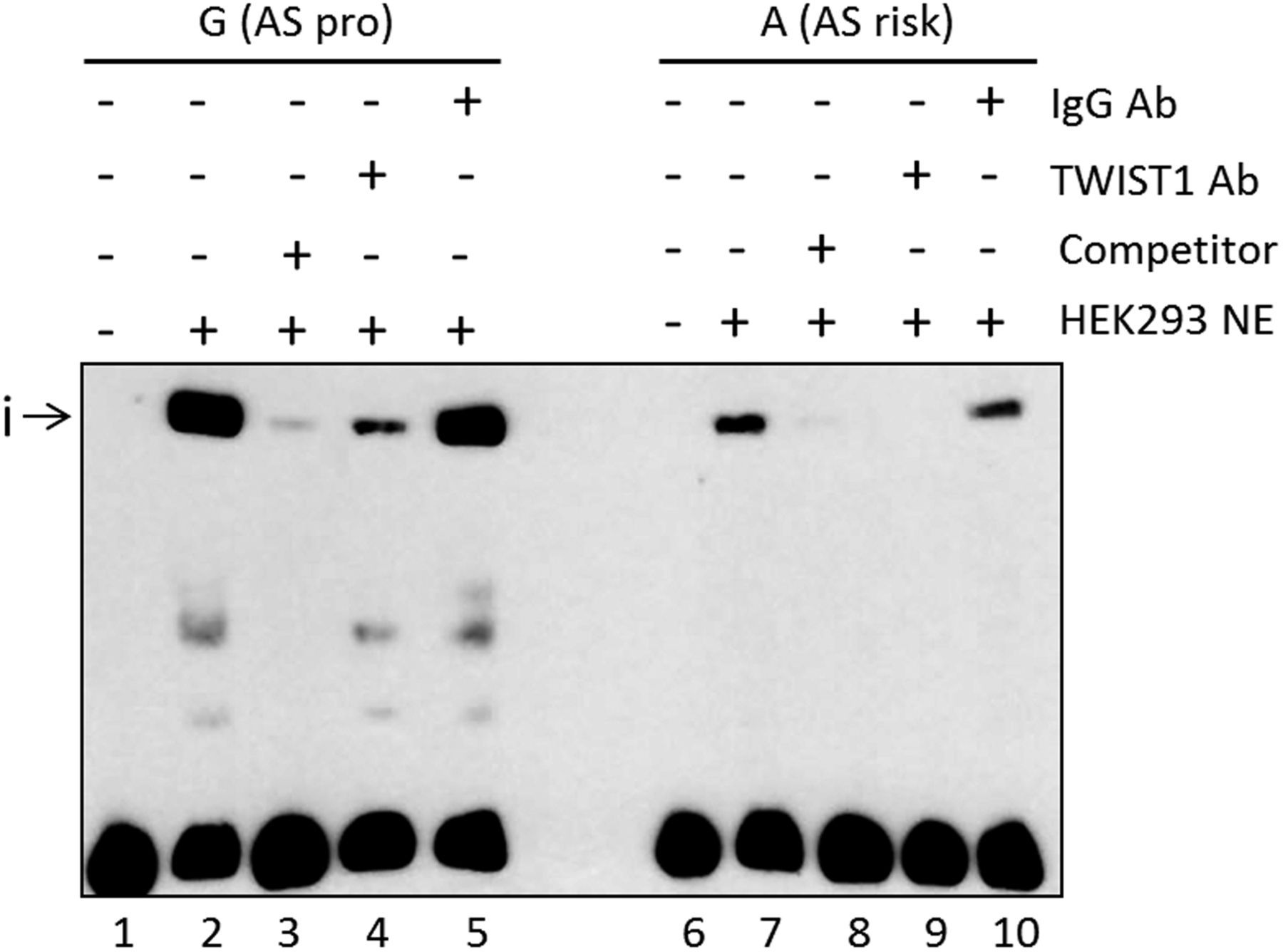
The ankylosing spondylitis (AS)-risk allele at rs11209032 alters DNA-protein complex formation. Chemiluminescent electrophoretic mobility shift assay (EMSA) showing complex formation (i) after addition of HEK293 nuclear extract (lanes 2 and 7), and competition with 100-fold excess of unlabelled probes (lanes 3 and 8). TWIST1 antibody addition leads to a reduction or inhibition of the complex (i) (lanes 4 and 9, respectively). Addition of nonspecific IgG antibody (lanes 5 and 10). NE, nuclear extract; pro, protective. This result was confirmed in five independent experiments.
A search for TF binding consensus sequences that directly overlap rs11209032 revealed a CTCF binding motif (in lymphoblastoid cell line GM12878).18 However, rs11209032 genotype had no influence on binding (data not shown). The consensus sequence of the Th1 transcriptional repressor TWIST1 (5′-NCANNTGN-3′) is located 1 bp 3′ of the rs11209032 SNP. HEK293 cells are a good source of TWIST1 protein (see online supplementary figure S3B). Addition of TWIST1 antibody to the EMSA reduced/inhibited the formation of the DNA-protein complex for both alleles (figure 3).
rs11209032 alters levels of H3K4me1 methylation
Publicly available data show that the region overlapping rs11209032 is enriched for enhancer-associated H3K4me1 methylation in Th1-cells.14 To investigate enhancer activity ex vivo, we assessed the levels of H3K4me1 methylation at rs11209032 by ChIP-qPCR. Homozygosity for the ‘A’ (risk) allele correlated with reduced H3K4me1 abundance in CD4+ T cells from patients with AS, whereas the level in ‘G/G’ homozygotes was similar to the positive control (IL10 enhancer) (figure 4A, p=0.02).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Homozygosity for the ankylosing spondylitis (AS)-risk allele at rs11209032 is associated with reduced enhancer activity without altering mRNA levels of IL23R or IL12RB2. (A) H3K4Me1 methylation ChIP-qPCR assessed at the rs11209032 locus in CD4+ T cells (PMA and ionomycin-stimulated) from three ‘G/G’ and three ‘A/A’ patients. The fold enrichment is expressed as mean±SEM for each patient in triplicate. Positive control is IL10 enhancer. Student's t test was used. (B) The transcriptional activity of rs11209032 compared with minP (set to 1) was measured by luciferase reporter assays in HEK293T cells. The values of relative luciferase activity are expressed as mean±SEM of three or four repeat experiments each done in triplicate. One-way analysis of variance was used. (C) Relative amount of IL23R mRNA in primary CD4+ T-cells (PMA and ionomycin-stimulated) from 6 GG and 6 AA patients (normalised against β-actin). mRNA levels are expressed as mean±SEM. Student's t test was used. (D) Relative amount of IL12RB2 mRNA in primary CD4+ T-cells (PMA and ionomycin-stimulated) from 6 GG and 6 AA patients (normalised against β-actin). mRNA levels are expressed as mean±SEM. Student's t test was used. minP, minimal promoter.
The AS-risk allele at rs11209032 shows reduced reporter activity
We used luciferase reporter assays to confirm the effect of rs11209032 genotype on enhancer activity. Luciferase reporter assays were performed in HEK293T cells transfected with pGL4.23 plasmids containing the minimal promoter (minP) and the 1.14 kb PRE sequence (see figure 1) with either the ‘A’ (risk) allele or the ‘G’ (protective) allele. The ‘G’ (protective) allele showed significantly increased reporter activity above the minP level of 1 (figure 4B, p<0.001), which provides further support to PRE having enhancer activity. Interestingly, the ‘A’ (risk) allele significantly reduced this activity compared with the protective allele (p<0.001).
No correlation between rs11209032 genotype and IL23R or IL12RB2 mRNA levels
We next assessed the effect of the rs11209032 genotype and reduced enhancer activity on expression levels of the nearby genes. Analysis of IL23R and IL12RB2 expression in CD4+ T-cells isolated from patients with AS (PMA and ionomycin-stimulated overnight, cell viability≥71%) with different rs11209032 genotypes (6 GG and 6 AA) showed no significant effect on mRNA levels (figure 4C,D). There was also no correlation between disease activity and mRNA levels for either gene (data not shown).
Discussion
We have clearly shown the primacy of the rs11209032 SNP in a potentially important regulatory region between IL23R and IL12RB2 associated with AS. The rs11209032 AS-risk ‘A’ allele influences the formation of a TF complex, which includes TWIST1. Luciferase reporter assays showed an effect of the AS-risk allele on transcription but, somewhat surprisingly, we could not detect an effect on expression of the two nearby genes IL23R or IL12RB2. Our experiments on gene expression were not highly powered and the absence of definitive high and/or low expresser cell type controls somewhat hindered their interpretation. Overall, these data suggest that this regulatory element does not influence the expression of the neighbouring genes. However, we have not formally excluded the possibility it could modulate the function of these genes under different stimulation conditions from those that we have used or according to the differentiation status of the cells. It is also possible that there may be other regulatory elements in this region, which we have not yet identified that could influence these genes. These possibilities will be investigated systematically in future studies.
AS is a complex polygenic disease showing multiple genetic associations with the IL-23 pathway. Here, we show that homozygosity for the AS-risk ‘A’ allele of an SNP in the IL-23R-IL12RB2 intergenic region is associated with increased Th1-cell numbers. We have not been able to define the regulatory mechanism(s) involved precisely. Our EMSA data show that TWIST1 binds to the region containing rs11209032. TWIST1 is reported to be a transcriptional repressor of Th1 gene expression and cytokine production, specifically reducing Ifn-γ expression in mouse cells.19 It is, therefore, possible that altered binding of TWIST1 to the putative enhancer may contribute to the findings reported here. We now intend to investigate more deeply the involvement of TWIST1 and the full nature of the DNA-protein complex with rs11209032, which is reduced in the presence of the risk ‘A’ allele, to look for a potential network of genes that might be regulated at least in part by this SNP.
Inspection of 2 Mb of flanking sequence around rs11209032 reveals no obvious Th1-related genes. However, since only 27% of distal regulatory elements are reported to interact with the nearest promoter, the target of an SNP disease association is often not with the nearest gene and may extend over several megabases.20 ,21 Substantial work may be required to identify the target gene(s).
Double-positive IL-17A+/IFN-γ+ CD4+ T-cells have been reported in synovial fluid and synovial tissue of some patients with rheumatoid arthritis (RA)22–24 and are increased in peripheral blood from patients with AS and RA.25 However, we found no evidence of an influence of SNP genotype on the frequency of these cells. Previously, increased IFN-γ production has been reported in T-cells from patients with AS,26–28 although this is controversial because others report reduced IFN-γ levels.29 Here, we report a correlation of rs11209032 genotype with the percentage of IFN-γ+ CD4+ T-cells in patients with AS. Our results highlight the potential importance of genetic/epigenetic regulation of the Th1 pathway in the pathogenesis of AS.
There are at least two distinct genetic effects arising from the vicinity of IL23R in AS. The first (rs11209026) has been previously characterised and encodes an Arg381Gln change in the cytoplasmic tail of IL-23R that reduces signalling both in healthy donors and patients with inflammatory arthritis.11 ,12 ,30 ,31 Such protein-coding changes constitute only a minority of genetic associations with common diseases, perhaps indicative of their relatively pronounced functional effects. In contrast, more subtle effects on gene expression or protein binding, as represented by the IL23R-IL12RB2 association we describe here, are probably more common and may act synergistically.32 Our study highlights the power of conditional analysis to identify the primary genetic disease associations definitively. On first inspection, rs6677188, which lies in the same associated region as rs11209032, appears somewhat more likely to be functionally relevant because it is closer to the local peak of DNase I hypersensitivity. However, the conditional analysis unequivocally demonstrated the primacy of the rs11209032 association since the apparent association with rs6677188 disappeared after conditioning on rs11209032.
Genetic associations with disease identified by GWAS can potentially identify new drug targets even where the strength of the association is relatively weak. In general, polymorphisms influencing gene expression are more likely to be implicated in polygenic diseases like AS. Additive influences arising from numerous SNPs in the IL-23 pathway, which alter the effector functions of Th1-cells and Th17-cells in patients with SpA, have been described32 but the full complexity of the regulation of these cells is only just becoming apparent.33
The combination of an analytical approach with a computational and experimental validation can identify important transcriptional gene networks, such as those governing the specification and function of Th1-cells in health and disease (eg, T-bet, STAT4, RUNX3 and TWIST1).19 ,34 A similar approach has been used to identify and explain the association between a functional polymorphism in the tumour necrosis factor receptor gene (TNFRSF1A) and multiple sclerosis.35 Further studies in AS will focus on identifying the factors contributing to the reduced enhancer activity reported here and how these factors contribute to increased Th1-cell signalling.
Acknowledgments
The authors wish to thank all the study participants who generously donated their DNA and peripheral blood to this study. The authors also thank Louise Appleton for DNA isolation of patient samples.
References
Footnotes
Handling editor Tore K Kvien
Contributors BPW and CJC are joint senior authors. ARR, MV, LC, AR, AC, JCK, PB, CJC and BPW: conceived and designed the experiments. ARR, LC, MV, AR and CJC: performed the experiments. ARR, MV, LC, PB, CJC and BPW: analysed the data. ARR, MV, LC, AC, JCK, PB, CJC and BPW: wrote the manuscript.
Funding ARR was funded by Arthritis Research UK (grant 20402), MV by National Institute for Health Research (NIHR) Oxford comprehensive Biomedical Research Centre (immunity and inflammation theme A93081), LC by Arthritis Research UK (grant 20235) and AR by NIHR Oxford Comprehensive Biomedical Research Centre. JCK is funded by the European Research Council under the European Union's Seventh Framework Programme (FP7/2007–2013)/ERC Grant agreement no. 281824, Arthritis Research UK (grant 20773) and the NIHR Oxford Comprehensive Biomedical Research Centre. Additional funding was provided by Arthritis Research UK (grants 19356, 18797 and 20796), the NIHR Thames Valley collaborative research network, NIHR Oxford Musculoskeletal Biomedical Research Unit and National Ankylosing Spondylitis Society (UK).
Competing interests None declared.
Ethics approval Oxford C Research Ethics Committee 06/Q1606/139 and Oxford B Research Ethics Committee 07/Q1605/35.
Provenance and peer review Not commissioned; externally peer reviewed.