Article Text

Abstract
Objectives A key clinical paradox in osteoarthritis (OA), a prevalent age-related joint disorder characterised by cartilage degeneration and debilitating pain, is that the severity of joint pain does not strictly correlate with radiographic and histological defects in joint tissues. Here, we determined whether protein kinase Cδ (PKCδ), a key mediator of cartilage degeneration, is critical to the mechanism by which OA develops from an asymptomatic joint-degenerative condition to a painful disease.
Methods OA was induced in 10-week-old PKCδ null (PKCδ−/−) and wild-type mice by destabilisation of the medial meniscus (DMM) followed by comprehensive examination of the histology, molecular pathways and knee-pain-related-behaviours in mice, and comparisons with human biopsies.
Results In the DMM model, the loss of PKCδ expression prevented cartilage degeneration but exacerbated OA-associated hyperalgesia. Cartilage preservation corresponded with reduced levels of inflammatory cytokines and of cartilage-degrading enzymes in the joints of PKCδ-deficient DMM mice. Hyperalgesia was associated with stimulation of nerve growth factor (NGF) by fibroblast-like synovial cells and with increased synovial angiogenesis. Results from tissue specimens of patients with symptomatic OA strikingly resembled our findings from the OA animal model. In PKCδ null mice, increases in sensory neuron distribution in knee OA synovium and activation of the NGF-tropomyosin receptor kinase (TrkA) axis in innervating dorsal root ganglia were highly correlated with knee OA hyperalgesia.
Conclusions Increased distribution of synovial sensory neurons in the joints, and augmentation of NGF/TrkA signalling, causes OA hyperalgesia independently of cartilage preservation.
- Knee Osteoarthritis
- Chondrocytes
- Fibroblasts
- Synovitis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Pain, a key reason for patients with osteoarthritis (OA) to seek medical assistance, diminishes quality of life and complicates healthcare management. OA is a common age-related degenerative joint disease in which structural damage to articular cartilage is a pathological hallmark. Pain perception in OA is linked to disease-modified nociception, rather than to cartilage destruction, because cartilage is normally not innervated.1 Inflammation of the synovial lining (synovitis) and bone marrow lesions are related to the severity of pain in OA.2 ,3 Yet, a fundamental clinical discrepancy remains—the degree of cartilage degeneration from radiographic and histological evidence does not correlate with perceived levels of pain sensation.4–6 Up to 40% of patients with severe radiographic changes in knee joints are symptom-free, indicating that joint pathology can be uncoupled from pain sensation.7 The mechanism by which OA develops from an asymptomatic condition to a painful disease represents a critical gap in our knowledge.
In this study, we explored possible mechanisms by which OA develops from an asymptomatic condition to a painful disease. Using an experimental OA model involving destabilisation of the medial meniscus (the DMM model) in mice,8 ,9 we comprehensively analysed the pathological role of protein kinase Cδ (PKCδ), a key mediator of cartilage degeneration.10–12 Our findings suggest that a lack of PKCδ protects cartilage from the OA-like pathological changes normally observed with DMM. This striking resistance to OA in PKCδ null mice clearly establishes that PKCδ signalling is required for the development of OA pathology and that the selective inhibition of PKCδ expression may, therefore, prevent cartilage degeneration. However, the absence of PKCδ exacerbated OA-associated pain. This OA-related hyperalgesia compelled us to characterise mechanisms by which PKCδ-mediated signalling induces OA pain independently of cartilage degeneration. The emerging molecular mechanism is that nerve growth factor (NGF) expression and tropomyosin receptor kinase (TrkA) expression by synovial fibroblasts, which is normally suppressed by PKCδ, is augmented during OA progression and promotes sensory nerve outgrowth of innervating dorsal root ganglia (DRGs).
Results
Loss of PKCδ protects mice from OA-like cartilage degeneration, but causes hyperalgesia
Our previous studies demonstrated that PKCδ is a rate-limiting kinase that activates cartilage catabolic pathways in cultured human articular chondrocytes.11–13 In the present study, we first examined histopathological changes in the knee joint following DMM surgery. Knee joints of PKCδ null mice showed resistance to cartilage degeneration compared with wild-type (WT) mice during OA progression after DMM surgery, as observed by histology (figure 1A), histopathology grading (figure 1B), gross microscopy (figure 1C) and μCT (figure 1D).

Genetic deletion of the protein kinase Cδ (PKCδ) gene in mice inhibits osteoarthritis (OA) pathogenesis but augments knee joint OA-associated hypersensitivity to pain. OA was induced by destabilisation of the medial meniscus (DMM) in 10-week-old PKCδ−/− and age-matched and gender-matched wild-type (WT) mice, followed by harvesting knee joints at 4 and 8 weeks post-DMM. Each knee shown is representative for a group of mice (n=12). (A) Histological assessment for proteoglycan depletion by Safranin-O fast green staining (×20). Arrows indicate degeneration of the articular cartilage surface. (B) Severity of articular cartilage degradation was graded using the Osteoarthritis Research Society International (OARSI) scoring system.14 Values are mean±SD (compared between WT and PKCδ−/−: *p<0.05; **p<0.01). (C) India ink staining: the damaged articular cartilage surface is clearly visible following DMM (arrows). (D) Architectural changes in subchondral bone analysed by μCT scanning. L, lateral; M, medial. (E) Development of mechanical allodynia (von Frey filament testing) in the ipsilateral hindpaw, comparing PKCδ−/− with age-matched and gender-matched WT mice following DMM. Values are mean±SD, p<0.01, F=12.8 (WT vs PKCδ−/−, each group: n=17). Sham groups (n=17 for each) show similar values in both WT and PKCδ−/−. ns, not significant. (F) Spontaneous rearing activity (vertical photobeam crossings), **p<0.01, and (G) ambulation (horizontal photobeam crossings) are reduced in PKCδ−/− compared with WT after DMM, *p<0.05. Values are mean±SD.
Unexpectedly, behavioural pain tests indicated that PKCδ null mice developed significantly lower pain thresholds (p<0.01) compared with WT mice after OA induction (figure 1E). Baseline pain tests showed no significant difference between WT and PKCδ null mice. This result suggests that development of OA pain can occur independently of the degree of knee cartilage degeneration. At 8 weeks post-DMM surgery, PKCδ null mice clearly developed movement-evoked pain as reflected by reduced spontaneous motor activity (figure 1F, G). Taken together, these behavioural results establish that loss of PKCδ sensitises mice to greater knee joint pain during OA development.
Increased density of peripheral nerves in OA synovium correlates with increased joint pain sensation
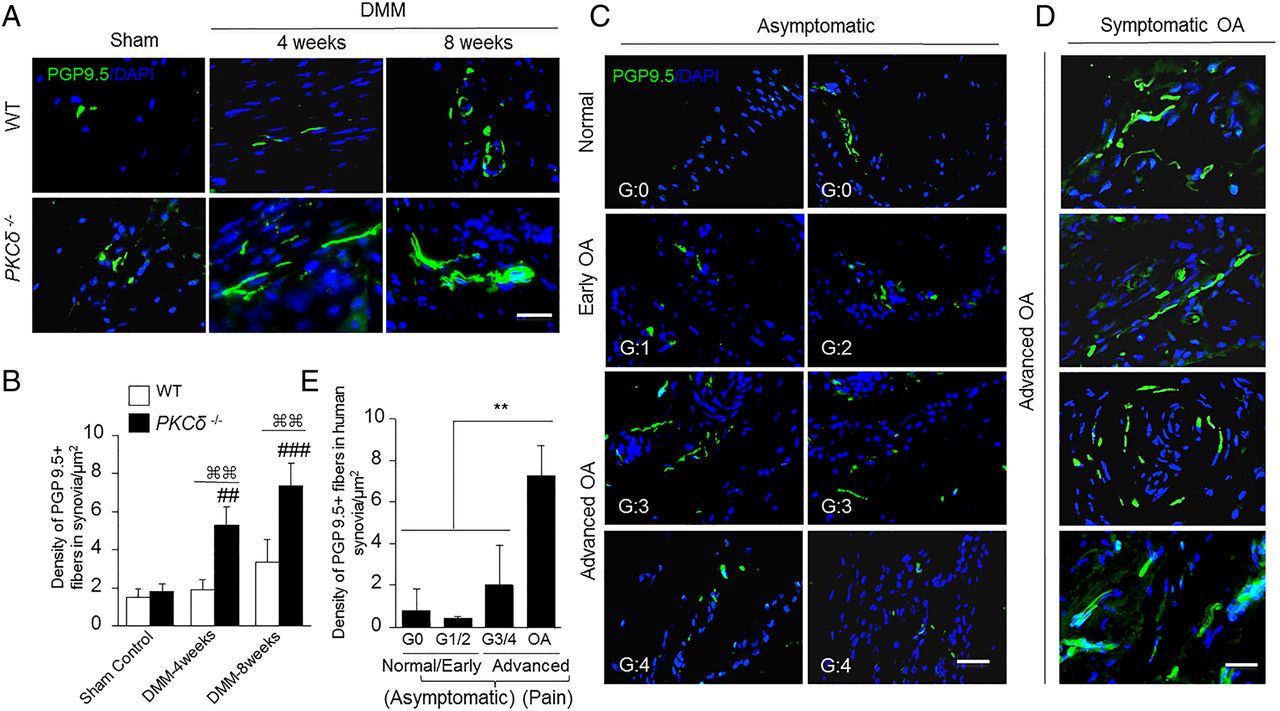
We investigated whether sensory nerve sprouting, a form of pathological neuroplasticity in the inflamed joints of geriatric mice,15 is a pathological feature of OA that contributes to the hyperalgesia. Peripheral nerve innervation was increased in the ipsilateral knee joint synovium of WT mice at 8 weeks post-DMM surgery, as shown by immunofluorescence staining with anti-PGP9.5 antibody. These PGP9.5-positive structures in knee joint synovium were significantly (p<0.01) higher in PKCδ null mice compared with WT mice (figure 2A, B).

Increased number of peripheral nerve fibres sprouting in osteoarthritis (OA) synovium correlates with increased joint pain sensation in OA. (A) Representative immunofluorescence images of staining for PGP9.5 (green) in knee joint synovium of PKCδ−/− and wild-type (WT) mice. (B) Quantitative analyses of nerve fibre sprouting. Following destabilisation of the medial meniscus (DMM) surgery, the density of PGP9.5 fibres in PKCδ−/− mice exhibits a significant increase in synovial/capsular regions compared with WT (⌘⌘p<0.01; values are expressed as mean±SD; n=5/group). At 4 and 8 weeks after DMM, PGP9.5 immunoreactivity in the synovia of PKCδ−/− mice showed increases compared with sham controls (## p<0.01; ###p<0.001), while the DMM-operated WT mice did not. (C and D) Human knee joint synovial and capsular tissues from asymptomatic organ donors with no history of chronic knee pain (G:0=normal; G:1/2=early OA; G:3/4=advanced OA) and surgically removed tissues from age-matched symptomatic OA subjects stained for PGP9.5 to detect nerve fibres sprouting. (E) Quantitative analyses showed no significant changes in neural distribution (PGP9.5+), regardless of cartilage degeneration stage (normal, early-OA or advanced-OA) in knee joint synovial specimens of age-matched asymptomatic organ donors. In contrast, a striking increase in nerve fibres (PGP9.5+) was detected in knee joint synovial specimens from symptomatic OA subjects compared with all groups of asymptomatic organ donors (**p<0.01; each group: n=3–5). PGP9.5+ nerve fibre density was calculated as the nerve fibre area divided by the total area examined (μm2/μm2). 4′,6-diamidino-2-phenylindole (DAPI) stains nuclei blue. G=OA grade. All scale bars, 50 μm.
We validated the findings of our animal studies by evaluating human knee joint specimens. Joint tissues were obtained from symptomatic OA subjects undergoing knee replacement surgery because of severe knee joint pain, as well as from organ donors with no history of joint disease or chronic knee joint pain. Asymptomatic knee joint synovial specimens showed no significant differences in neural fibre distribution (PGP9.5+) among the different grades of cartilage degeneration (figure 2C). However, nerve fibre innervation was markedly increased in knee synovial specimens from patients with symptomatic OA (figure 2D, E). These results show that the degree of OA pain robustly correlates with increased peripheral nerve fibres in human knee joints.
OA-induced hyperalgesia is associated with increased NGF/TrkA signalling
The pronounced induction of peripheral nerve sprouting during development of OA hyperalgesia led us to investigate PKCδ-regulated axonal growth promoting factors that stimulate neuronal sprouting. Deletion of PKCδ using siRNA in human synovial cells selectively upregulated expression of NGF/TrkA, brain-derived nerve growth factor and Substance P (tachykinin; TAC1)—downstream targets of the NGF/TrkA axis16 ,17 (see online supplementary figure S1A, B). Deletion of PKCδ in chondrocytes did not change the expression of any of these factors (see online supplementary figure S1C, D). Corroborating the results of these mechanistic studies, the genetic loss of PKCδ in mice significantly enhanced synovial expression levels of NGF/TrkA during OA progression (4 and 8 weeks post-DMM) compared with WT mice (figure 3A–D). In articular chondrocytes, the NGF level was modestly increased in advanced OA (8 weeks post-DMM) in both WT and PKCδ null mice (see online supplementary figure S2A, B). Abundant basal expression of TrkA was also observed in chondrocytes, without significant changes during OA progression in both WT and PKCδ null mice (see online supplementary figure S2C, D). We examined whether stimulation of NGF expression in the painful OA knee joint could result from immune cell infiltration (eg, macrophages). Immunofluorescence staining showed increased CD11b-positive cells at 4 and 8 weeks post-DMM in WT mice. In contrast, CD11b-positive cells were significantly diminished in PKCδ null mice (see online supplementary figure S3).

Osteoarthritis (OA)-induced hyperalgesia is associated with increased nerve growth factor (NGF)/tropomyosin receptor kinase (TrkA) axis signalling in knee synovium. (A) Representative immunofluorescence staining for NGF (green) in the knee joint synovium of PKCδ−/− and wild-type (WT) mice. n=5/group. (B) Quantitative analysis of NGF expression in the synovium. (C) Immunofluorescence staining for TrkA (green) in knee joint synovium of PKCδ−/− and WT mice. (D) Quantitative analysis of TrkA expression in synovium. For all quantitative analyses, values are mean±SD (compared between WT sham and destabilisation of the medial meniscus (DMM): *p<0.05, **p<0.01; compared between PKCδ−/− mice sham and DMM: #p<0.05, ##p<0.01, ###p<0.001; compared between WT and PKCδ−/: ⌘⌘p<0.01). (E) Representative immunofluorescence images show significantly increased expression of NGF (green) in synovium of symptomatic OA subjects. Arrows indicate NGF-expressing nerve endings. n=6 per group. (F) Quantitative analyses of NGF expression in human synovium. Values are mean±SD (compared between with G:0–4: ***p<0.001). (G) Representative immunofluorescence images of TrkA (red) expression in human synovium. Each group: n=6. (H) Quantitative analysis of TrkA expression in human synovium. Values are presented as mean±SD (compared with G:0: *p<0.05, ***p<0.001; compared with G:3 and G:4: ##p<0.01). 4′,6-diamidino-2-phenylindole (DAPI) stains nuclei blue. G=OA grade. All scale bars, 50 μm. FI, fluorescence intensity; a.u., arbitrary unit.
We next assessed the activity of PKCδ in human joint synovium. Strikingly, the activity of PKCδ was significantly decreased in synovia of symptomatic patients (see online supplementary figure S4). In contrast, the level of NGF was highly elevated at joint synovia from patients with symptomatic OA, but not in specimens from asymptomatic organ donors (figure 3E, F). Expression levels of TrkA were moderately increased during OA progression in the asymptomatic group (figure 3G, H). Similar to NGF, TrkA levels were strongly increased in symptomatic OA synovium compared with the asymptomatic group. In the synovium from patients with symptomatic OA, we found increased co-localisation of NGF expression with peptidergic sensory nerve fibres (calcitonin gene-related peptide-positive), suggesting active axonal growth activity in painful knee joints (see online supplementary figure S5A). Double labelling for NGF and CD68, a marker for immune cell infiltration (eg, macrophages), revealed that markedly increased levels of NGF and moderately increased levels of CD68 were evident in knee synovial tissues from patients with symptomatic OA. Only a few NGF-positive cells co-localised with CD68 (see online supplementary figure S5B), suggesting that fibroblast-like synovial cells are a dominant source of NGF production in the OA knee joint.
In human chondrocytes, however, both NGF and TrkA were abundantly expressed at all stages of cartilage degeneration. NGF levels were low in normal cartilage from subjects with early stages of OA, but increased in cartilage from subjects with advanced stages of OA. There was no difference in NGF expression between cartilage from symptomatic and asymptomatic individuals (see online supplementary figure S6A, B). The expression of TrkA did not differ significantly among different pathological grades of cartilage (see online supplementary figure S6C, D).
PKCδ deletion inhibits production of inflammatory cytokines and cartilage degrading enzymes
PKCδ expression is activated in human articular chondrocytes by pro-inflammatory cytokines, such as tumour necrosis factor-α (TNFα) or interleukin-1β (IL-1β), and by cartilage breakdown products, such as fibronectin fragments, to elicit stimulation of catabolic enzymes.11 ,13 We explored the nature of this anti-inflammatory effect in PKCδ null mice using whole joint extracts collected at 4 and 8 weeks post-DMM and quantitative PCR (qPCR). Expression of multiple inflammatory mediators and cartilage degrading enzymes were strongly suppressed in PKCδ null mice (see online supplementary table S1).
To identify the tissue source of inflammatory cytokine production, we examined expression levels of TNFα and IL-1β—two representative pro-inflammatory cytokines in OA pathology.18 Expression levels of TNFα (figure 4A–C) and IL-1β (figure 4D–F) were greatly increased in cartilage and synovium of WT mice at 4 and 8 weeks post-DMM. Importantly, these elevated levels were not evident in the joints of PKCδ null mice. Consistent with the results from our murine OA model, expression levels of TNFα (figure 4G–I) were induced in both cartilage and synovium during human knee OA progression. The levels of TNFα showed a robust correlation with joint pathology, but not with the level of pain.

Deletion of the protein kinase Cδ (PKCδ) gene in mice suppresses expression of inflammatory cytokines in the knee joint following destabilisation of the medial meniscus (DMM) surgery. (A) Representative immunofluorescence images of tumour necrosis factor-α (TNFα) (green) expression in cartilage and synovium of PKCδ−/− and wild-type (WT) mice. Each group: n=5. (B and C) Quantitative analyses of TNFα expression in cartilage and synovium. Values are mean±SD (compared between WT sham and DMM: **p<0.01; compared between PKCδ−/− mice sham and DMM: #p<0.05; compared between WT and PKCδ−/−: ⌘p<0.05). 4′,6-diamidino-2-phenylindole (DAPI) stains nuclei blue. (D) Representative immunofluorescence images of interleukin (IL)-1β expression (green) in cartilage and synovium of PKCδ−/− and WT mice. Each group: n=5. (E and F) Quantitative analyses of IL-1β expression in cartilage and synovium. Values are mean±SD (compared between WT sham and DMM: **p<0.01; compared between PKCδ−/− mice sham and DMM: #p<0.05; compared between WT and PKCδ−/−: ⌘p<0.05). (G) Immunohistochemical analyses of TNFα in human cartilage and synovium. Positive cells stain brown, Nuclei are counterstained with haematoxylin (blue) (top row). DAPI stains nuclei (blue) (bottom row). Each group: n=6. (H and I) Quantitative analyses of TNFα expression in human articular cartilage and synovium. Values are mean±SD (compared with G:1: *p<0.05; **p<0.01). G=osteoarthritis (OA) grade. All scale bars, 50 μm.
Loss of PKCδ augments knee joint angiogenesis
Previously, our group19 and others20 ,21 demonstrated that experimental OA in mice leads to pathological angiogenic features that mimic those seen in OA patients with chronic joint pain. Induction of OA by DMM surgery resulted in progressive angiogenesis in joint synovium at 4 and 8 weeks post-DMM, as reflected by increases in (i) the endothelium marker CD31 (figure 5A, B) and (ii) the potent angiogenic factor, vascular endothelial growth factor (VEGF) (figure 5C, D). Compared with WT mice, aggressive angiogenic activity developed in PKCδ null mice with overt structural remodelling of the vasculature in the knee synovial joint after DMM surgery.

Augmented angiogenesis in knee joints of PKCδ−/− mice following destabilisation of the medial meniscus (DMM) surgery. (A and C) Representative immunofluorescence images show significantly increased expression of both CD31 (green) (A) and vascular endothelial growth factor (VEGF) (green) (C) expression in the knee joint synovium of PKCδ−/− mice after DMM compared with wild-type (WT) mice. Arrows indicate vascularisation. Each group: n=5. (B and D) Quantitative analysis of CD31 and VEGF expression in synovium. Values are mean±SD (compared between WT sham and DMM: *p<0.05; **p<0.01; compared between PKCδ−/− mice sham and DMM: ##p<0.01; compared between WT and PKCδ−/−: ⌘p<0.05. (E) Representative immunofluorescence images show increased ATF4 expression (green) in synovium of PKCδ−/− mice compared with WT mice. Each group: n=5. (F) Quantitative analyses of activating transcription factor-4 (ATF4) expression in synovium. Values are mean±SD (compared between WT sham and DMM: *p<0.05; compared between PKCδ−/− mice sham and DMM: #p<0.05; compared between WT and PKCδ−/−: ⌘p<0.05). (G–J) qPCR analyses of ATF4 and VEGF genes in human fibroblast-like synovial cells and mouse endothelial cells after knockdown of PKCδ expression by siRNA. Values are mean±SD (compared with control: *p<0.05; **p<0.01). 4′,6-diamidino-2-phenylindole (DAPI) stains nuclei blue. All scale bars, 50 μm.
We investigated whether loss of PKCδ genes induces increases in activating transcription factor-4 (ATF4) expression—a transcriptional factor that regulates angiogenesis.22 The ATF4 expression level was significantly increased (p<0.05) in the joint synovium during OA progression at (4 and 8 weeks post-DMM) in PKCδ null mice (figure 5E, F). Moreover, we found that introduction of siRNA for PKCδ into either human endothelial or synovial cells stimulated the expression of both ATF4 and VEGF (figure 5G–J). There were no changes in the levels of ATF4 or VEGF following introduction of siPKCδ into human articular chondrocytes (see online supplementary figure S1E).
PKCδ deficiency increases stimulation of the NGF/TrkA axis in DRG sensory neurons
The development of chronic OA pain is associated with neuronal plasticity that transmits pain signals that involve alterations in gene expression in DRG sensory neurons.19 ,23 Upon induction of knee OA pain in mice at 4 and 8 weeks post-DMM, we isolated ipsilateral L3–5 DRGs and did biochemical and molecular analyses. We found a striking suppression of PKCδ expression in the DRG neurons of WT mice during OA progression at 4 and 8 weeks post-DMM as assessed by qPCR (figure 6A) and immunofluorescence staining (figure 6B). In DRG neurons, expression levels of NGF and TrkA were greatly augmented in PKCδ null mice compared with WT mice, as assessed by qPCR (figure 6C, D) as well as by double immunofluorescence staining (figure 6E, F). NGF activates extracelluar signal regulated kinase/mitogen activated protein kinase (ERK/MAPK) and sustained ERK activation plays a key role in centralisation and maintenance of chronic pain.24 Our immunofluorescence staining data show that ERK activation was increased under chronic OA pain conditions in the DRG neurons of WT mice, and this activation was clearly increased in PKCδ null mice during OA progression (see online supplementary figure S7A–C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Increased expression of the nerve growth factor (NGF)/tropomyosin receptor kinase (TrkA) axis and retrograde transport of NGF in dorsal root ganglia (DRG) following destabilisation of the medial meniscus (DMM) surgery. (A) Quantitative PCR (qPCR) analyses of ipsilateral L3–5 DRGs of PKCδ−/− and wild-type (WT) mice at 4 and 8 weeks post-DMM. Values are mean±SD (compared with sham control, **p<0.01). (B) Double immunofluorescence staining of protein kinase Cδ (PKCδ) (green) and MAP2 (red) show dramatically reduced PKCδ immunoreactivity in the DRG neurons of WT mice during osteoarthritis (OA) progression. Note that no PKCδ is detected in the DRGs of PKCδ−/− mice (lower row). PKCδ is only expressed in small-sized and medium-sized afferent neurons in the DRG. Each group: n=5. (C and D) qPCR analysis of NGF (C) and TrkA (D) expression levels in L3–5 DRGs. Values are mean±SD (compared with sham control: *p<0.05). Double immunofluorescence staining of NGF (green) (E) or TrkA (green) (F) and NeuN (red) in DRGs of PKCδ−/− and WT mice. Co-localisation of the two stains appears yellow. n=5 for each group. (G) Representative immunofluorescence images of retrograde transport of NGF-Biotin (green) from peripheral sensory neuronal terminals in knee joint to the soma of L3–5 DRG neurons. NeuN (red) is a marker of neurons. Arrows indicate DRG neurons positive for NGF-Biotin. Anti-NGF antibody administration abolished retrograde NGF-Biotin transport to L3–5 DRGs. 4′,6-diamidino-2-phenylindole (DAPI) stains nuclei blue. Each group: n=5. All scale bars, 100 μm. (H) von Frey filament testing in the ipsilateral hindpaw, comparing PKCδ−/− mice receiving twice a week anti-NGF-2.5S antibody (30 μg in 5 μL saline) or saline (vehicle) till 8 weeks post-DMM surgery (this administration schedule is indicated by black arrows). PKCδ−/− mice injected with anti-NGF antibody showed significantly reduced pain compared with the saline control group from the third week of anti-NGF antibody administration. Sham-operated mice with saline injections were used as controls. Values are mean±SD (compared between PKCδ−/− mice with anti-NGF antibody and PKCδ−/− mice with saline: *p<0.01). For each group: n=10.
We next determined whether OA-induced augmentation of NGF levels in synovium leads to retrograde transport of NGF to DRG neuron cell bodies from innervating nerve terminals in the knee joint synovium. It is known that NGF is internalised as an NGF-TrkA complex and is retrogradely transported from peripheral terminals to sensory cell bodies in the DRG, where it activates transcription of a number of genes related to chronic pain.25 ,26 Our immunofluorescence results show significant retrograde transport of NGF-Biotin in the soma of L3–5 DRG neurons in PKCδ null mice compared with WT mice (figure 6G). Moreover, retrograde transport of NGF-Biotin was substantially abolished in both PKCδ null and WT mice intra-articularly injected with anti-NGF antibody. Behavioural pain tests also showed significantly reduced OA-induced pain in PKCδ null mice injected with anti-NGF antibody compared with a saline injection control group (figure 6H).
Discussion
The mechanism by which OA develops from an asymptomatic condition to a painful disease has been a critical gap in our knowledge. The complementarity between our results from an experimental OA animal model and our human subject findings has significant ramifications for resolving a long-standing clinical discrepancy of the lack of correlation between histological damage and the degree of joint pain. We now show that the PKCδ is a key inhibitor of NGF/TrkA axis signalling and sensory nerve sprouting in synovial tissues and DRGs, and thus alleviates OA hyperalgesia. This beneficial function of PKCδ in reducing synovial innervation is diametrically opposed to the inflammation-responsive catabolic function of PKCδ in articular chondrocytes that mediates cartilage destruction. We postulate that the degree of pain in OA is directly attributable to changes in PKCδ signalling in sensory neurons rather than to joint pathology per se. Therefore, understanding the neural mechanisms by which PKCδ controls neuronal signalling pathways in the knee joint is important for the development of clinical treatments to negate pain-related symptoms in patients with OA. Our study demonstrates that neurite outgrowth and the NGF-TrkA axis are important drivers for OA hyperalgesia in experimental OA in mice and symptomatic knee OA in humans, independent of the degree of cartilage degeneration (see online supplementary figure S8). A key finding of our work is that PKCδ prevents synovial augmentation of sensory fibres by attenuating signalling through the suppression of the NGF/TrkA axis in DRG neurons. Our results provide a mechanistic understanding of the findings of McNamee and colleagues,27 who elegantly demonstrated that induction of NGF expression in the joint correlates with pain-related behavioural changes during development of experimental OA.
The role of NGF in joint destruction is not clear. Our findings suggest that the NGF/TrkA axis may have a distinct regulatory role in chondrocytes and support cartilage homeostasis. First, NGF and TrkA are abundantly expressed in normal and healthy joint tissues, notably in chondrocytes and chondrocyte-like meniscus cells (unpublished data), in both WT and PKCδ null mice. Second, the NGF/TrkA axis is highly stimulated in the synovium of PKCδ null mice, which are strongly resistant to cartilage degeneration. Our results strongly suggest that fibroblast-like synovial cells, chondrocytes and meniscus cells, are primary sources of NGF, and possibly other pro-inflammatory cytokines and chemokines in OA joints. Our findings complement previous studies that indicate that NGF and chemokines are produced by inflamed fibroblast-like synovial cells and human OA chondrocytes.28–30 In addition, an increase in levels of NGF in human OA may promote angiogenesis and osteochondral vascularity.31 Based on these collective findings, it appears that increased amounts of NGF detected in arthritic synovial fluid32 could result from the cumulative levels of NGF secreted from several distinct joint tissues. Despite the encouraging efficacy of pharmacological NGF blockade in the clinical treatment of OA pain in patients,33 significant adverse effects of such blockade on joint integrity necessitate further studies on the biological actions of NGF in joint pathology.
Angiogenesis promotes ingrowth of sensory neurons into peripheral knee joint tissues exposed to damage and can contribute to persistent pain even after inflammation has subsided.34 Angiogenic features in experimental OA resemble those pathological changes seen in OA patients with chronic joint pain.19–21 We found significantly increased angiogenic activity in PKCδ null mice (compared with WT mice) during OA progression. Hence, angiogenic events may be involved in OA-associated pain sensation rather than solely in joint pathology. Although NGF is increased in human OA and may promote angiogenesis and osteochondral vascularity,35 we did not find appreciable induction of subchondral neovascularisation at 4 and 8 weeks post-DMM (unpublished data); yet, we note that an overt angiogenic event in joints at later stages (16 weeks post-DMM) cannot be excluded.
NGF activates ERK/MAPK and sustained ERK activation plays a key role in centralisation and maintenance of chronic pain.24 Our results demonstrate that, during OA progression, OA-induced pain diminishes PKCδ expression while it activates ERK and the NGF/TrkA axis in innervating L3/L5 DRG neurons of WT mice. This activation of ERK and NGF/TrkA axes was strikingly augmented by the absence of PKCδ in our null mice, suggesting that PKCδ signalling negatively regulates the NGF/TrkA-ERK axis in sensory neurons; the lack of PKCδ signalling in DRG neurons leads to overexpression of NGF and TrkA. The resulting induction of NGF/TrkA signalling promotes axonal outgrowth and increases sensory fibres in the joint, which demonstrates a very strong correlation between NGF-ERK activation and OA-induced hyperalgesia during OA progression.
In summary, we show that synovial sensory neurons in the OA joint and the NGF/TrkA axis are mechanistically linked to OA hyperalgesia. Thus, the inhibition of axonal outgrowth by targeting synovial levels of axonal outgrowth promoting factors, particularly NGF and TrkA, may represent an effective strategy for mitigating OA-associated pain.
Methods
Animals
Male and female 10-week old PKCδ null mice and WT mice with a C57BL/6 background were used for the animal experiments.
Human tissues
Details of the human tissues are described in online supplementary table S2.
Induction of OA in mice
OA was induced by DMM as we previously described.9 For additional details, see online supplementary methods.
Animal behavioural tests
Animal behavioural tests were done as we previously described.36 For additional details, see online supplementary methods.
Histology, macroscopic imaging, immunohistochemistry and histomorphometry
Histological and immunohistochemical analyses were performed as we previously described.37 Gross knee-joint pathology and µCT analyses were performed using standard procedures described previously.19 For additional details, see online supplementary methods.
Human primary cell isolation, culture and siRNA transfection
Human primary cells were cultured and transiently transfected with validated PKCδ siRNA as previously described.38 ,39 For additional details, see online supplementary methods.
Reverse transcription and qPCR analyses
Reverse transcription and qPCR were performed using standard procedures. For additional details, see online supplementary methods.
Intra-articular anti-NGF-2.5S injection and retrograde NGF-biotin transport
PKCδ null and WT mice with DMM or sham surgery received intra-articular injections of anti-NGF-2.5S antibody or mNGF2.5S-Biotin for retrograde studies. For additional details, see online supplementary methods.
Statistical analysis
Statistical significance was determined by Student's t test or analysis of variance for repeated measures, followed by step-down Bonferroni's multiple comparison post-test, as appropriate, using SPSS V.17 software (IBM Corporation). p Values <0.05 were considered to be statistically significant.
Acknowledgments
The authors thank the Gift of Hope Organ Tissue Donor Network as well as Drs Cs-Szabo, Chubinskaya and Margulis for making human tissues available. They also extend their appreciation to the family members of the tissue donors who made donations possible. The authors are grateful to Dr Keiichi Nakayama for PKCδ null mice. They also thank Dr Hazel Lum for her generous gift of human primary endothelial cells.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
Handling editor Tore K Kvien
RKc and XL contributed equally and are joint first authors.
Contributors RK and XL contributed equally. RK performed experiments, analysed data and wrote the manuscript. XL, JSK, ZL, JL and JLH performed experiments and analysed data. DC, GX, BL, AJvW, MP, DAS, DB and EK analysed data. H-JI designed experiments, analysed data and wrote the manuscript. All authors reviewed the manuscript.
Funding This work was supported by an NIH NIAMS R01 grant (AR053220) to HJI; an NIH NIAMS R01 grant (AR062136) to HJI; an R21 grant (AR067935) to HJI; a VA BLD&R Merit Review Award to HJI and an Arthritis Foundation to HJI.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Human Investigation Committee of Rush University Medical Center.
Provenance and peer review Not commissioned; externally peer reviewed.