Article Text

Abstract
Objectives To evaluate the long-term efficacy and safety of subcutaneous (SC) tocilizumab (TCZ) versus intravenous (IV) TCZ, including switching formulations, in patients with rheumatoid arthritis (RA) and inadequate response to disease-modifying antirheumatic drugs (DMARDs).
Methods Patients (n=1262) were randomised 1:1 to receive TCZ-SC 162 mg weekly (qw)+placebo-IV every four weeks (q4w) or TCZ-IV 8 mg/kg q4w+placebo-SC qw in combination with DMARD(s). After a 24-week double-blind period, patients receiving TCZ-SC were re-randomised 11:1 to TCZ-SC (n=521) or TCZ-IV (TCZ-SC–IV, n=48), and patients receiving TCZ-IV were re-randomised 2:1 to TCZ-IV (n=372) or TCZ-SC (TCZ-IV–SC; n=186). Maintenance of clinical responses and safety through week 97 were assessed.
Results The proportions of patients who achieved American College of Rheumatology (ACR)20/50/70 responses, Disease Activity Score in 28 joints remission and improvement from baseline in Health Assessment Questionnaire Disability Index ≥0.3 were sustained through week 97 and comparable across arms. TCZ-SC had a comparable safety profile to TCZ-IV through week 97, except that injection site reactions (ISRs) were more common with TCZ-SC. Safety profiles in patients who switched were similar to those in patients who received continuous TCZ-SC or TCZ-IV treatment. The proportion of patients who developed anti-TCZ antibodies remained low across treatment arms. No association between anti-TCZ antibody development and clinical response or adverse events was observed.
Conclusions The long-term efficacy and safety of TCZ-SC was maintained and comparable to that of TCZ-IV, except for ISRs. Profiles in patients who switched formulations were comparable to those in patients who received TCZ-IV or TCZ-SC. TCZ-SC provides additional treatment options for patients with RA.
Trial registration number NCT01194414.
- Rheumatoid Arthritis
- DMARDs (biologic)
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic, progressive disease associated with inflammation and degeneration of the joints and surrounding tissue. Treatments that have demonstrated efficacy with a favourable safety profile, such as antitumor necrosis factor inhibitors (aTNFs), an interleukin 6 (IL-6) receptor antagonist, a Janus kinase inhibitor and a T-cell co-stimulator are available.1 However, when faced with multiple choices, patients have demonstrated a preference for subcutaneous (SC) over intravenous (IV) therapies.2 ,3
Tocilizumab (TCZ) is a recombinant humanised monoclonal antibody directed against the human IL-6 receptor. Intravenous TCZ (TCZ-IV) is approved in over 100 countries and has demonstrated efficacy with a well-established safety profile in patients with RA.4–9 Subcutaneous TCZ (TCZ-SC) has demonstrated efficacy with a similar safety profile as TCZ-IV10–13 and has been approved in the USA, Switzerland, Canada, Japan, and the European Union.
The phase III SUMMACTA study evaluated the efficacy and safety of TCZ-SC in combination with disease-modifying antirheumatic drugs (DMARDs) in patients with moderate-to-severe RA and inadequate response to ≥1 DMARD.10 SUMMACTA met its primary endpoint and demonstrated non-inferiority of TCZ-SC 162 mg weekly (qw) to TCZ-IV 8 mg/kg every 4 weeks (q4w) with regard to the American College of Rheumatology 20 (ACR20) response at week 24. TCZ-SC had a safety profile comparable to TCZ-IV.10 To assess the long-term efficacy and safety of TCZ-SC, data up to week 97 from the SUMMACTA study are evaluated here.
Patients and methods
Participants
Patient selection criteria were previously described.10 All patients had an inadequate response to ≥1 DMARD, and ≤20% had an inadequate response to aTNF(s). Patients continued to receive ≥1 non-biological DMARD at the stable pre-entry dose.
Study design
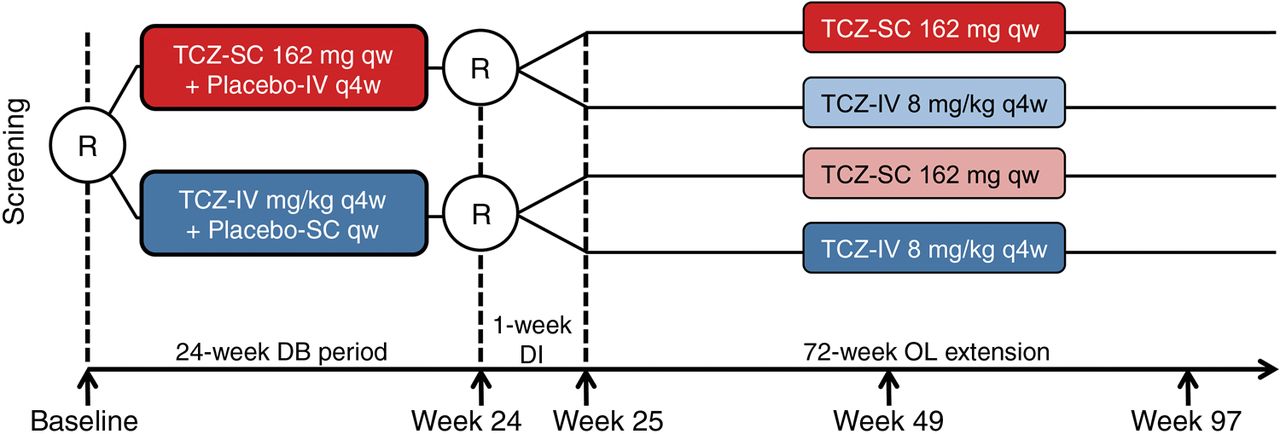
SUMMACTA was a 97-week, randomised, double-dummy, active-controlled, parallel-group, multicentre phase III trial with a 24-week double-blind (DB) period followed by a 72-week open-label (OL) extension, with a 1-week dose interruption at week 24 before the first OL dosing at week 25 (figure 1). Patients were stratified by geographic region and body weight (<60, 60 to <100 or ≥100 kg). At baseline, patients were randomised 1:1 to receive TCZ-SC 162 mg qw+placebo-IV q4w or TCZ-IV 8 mg/kg q4w+placebo-SC qw for the 24-week DB period. At week 24, patients in the TCZ-SC arm were re-randomised 11:1 to either continue to receive TCZ-SC or switch to TCZ-IV (TCZ-SC–IV), whereas patients in the TCZ-IV arm were re-randomised 2:1 to receive either TCZ-IV or TCZ-SC (TCZ-IV–SC).
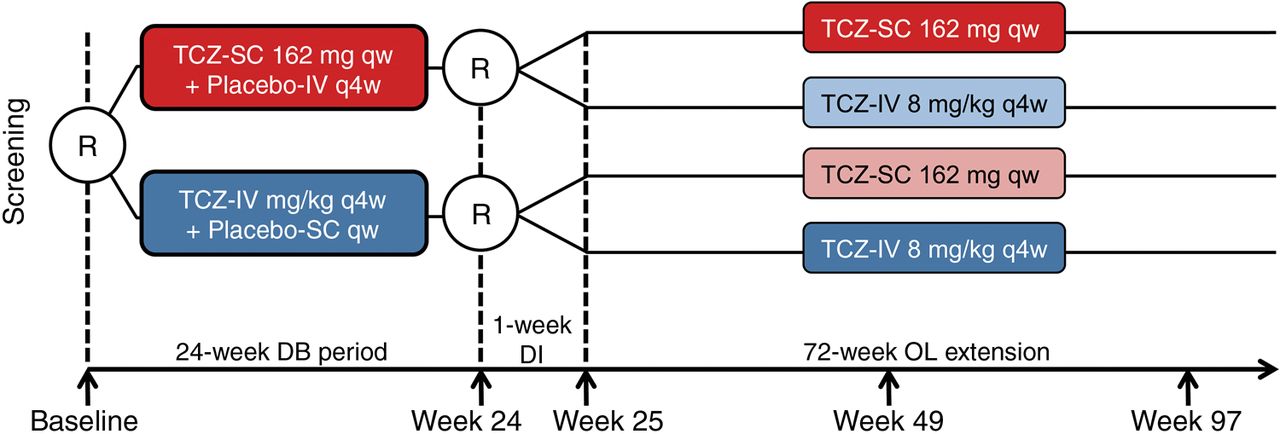
Study design. All patients continued to receive ≥1 permitted traditional disease-modifying antirheumatic drug at the stable pre-entry dose as prescribed by the treating physician. DB, double-blind; DI, dose interruption; IV, intravenous; OL, open-label; qw, weekly; q4w, every four weeks; R, randomisation; SC, subcutaneous; TCZ, tocilizumab.
Outcomes and assessments
Efficacy was assessed in the DB period at weeks 2, 4 and every four weeks thereafter and in the OL period at weeks 37, 49, 73 and 97 and/or at early withdrawal. Clinical assessments included ACR20/50/70; Disease Activity Score in 28 joints using erythrocyte sedimentation rate (DAS28-ESR) remission (DAS28 <2.6) and DAS28-ESR low-disease activity (LDA; DAS28 <3.2); Clinical Disease Activity Index (CDAI) remission (CDAI ≤2.8) and Boolean remission; withdrawal due to lack of therapeutic response; Health Assessment Questionnaire Disability Index (HAQ-DI) response (HAQ-DI score decreases of ≥0.3); and changes in HAQ-DI score. TCZ serum concentration was monitored for pharmacokinetic (PK) analysis. Pharmacodynamic (PD) parameters included C reactive protein (CRP) level and ESR.
Safety and immunogenicity
Safety was assessed throughout the study and for 8 weeks after week 97 or early withdrawal. Assessments included adverse events (AEs) and laboratory parameters at weeks 2, 4 and every four weeks thereafter. Anti-TCZ antibodies were assessed as previously described.10
Statistical methods
The intent-to-treat (ITT) population was used for all laboratory analyses and included patients who completed 24 weeks of DB treatment, were re-randomised at week 24 and received ≥1 TCZ dose after re-randomisation. Efficacy analyses included patients who also had a valid assessment at the time point analysed (completer analysis).
The ITT-PK population included all patients eligible for the ITT population who provided ≥1 sample evaluable for PK analysis. The safety population included all patients who received ≥1 dose of study drug and had ≥1 postdose safety assessment. Subgroup analyses for DAS28 remission and safety were conducted in all body weight categories.
Results
Patient disposition and baseline characteristics
A total of 1136 patients completed week 24 and 1135 entered the OL period (figure 2). The ITT population consisted of 521 patients in the TCZ-SC arm, 372 in the TCZ-IV arm, 186 in the TCZ-IV–SC arm and 48 in the TCZ-SC–IV arm. During the OL period, safety-related withdrawals occurred in 37 patients (7.1%) in the TCZ-SC arm, 24 (6.5%) in the TCZ-IV arm, 14 (7.5%) in the TCZ-IV–SC arm and 2 (4.2%) in the TCZ-SC–IV arm.

Patient disposition over 97 weeks. All patients received ≥1 dose of study drug and were eligible for inclusion in the intent-to-treat (all patients who received ≥1 dose of study drug) and safety (all patients who received ≥1 dose of TCZ and had ≥1 postdose safety assessment) populations. AE, adverse event; IV, intravenous; OL, open-label; qw, weekly; q4w, every 4 weeks; SC, subcutaneous; TCZ, tocilizumab.
Baseline demographics and clinical characteristics were balanced across arms (see online supplementary table S1); exceptions were the proportions of patients positive for anticitrullinated protein antibody (ACPA) and rheumatoid factor, which were lower in the TCZ-SC–IV arm than the TCZ-SC arm and higher in the TCZ-IV–SC arm than the TCZ-IV arm. Additionally, patients who received previous aTNFs were higher in the TCZ-SC–IV arm than the TCZ-SC arm and lower in the TCZ-IV–SC arm than the TCZ-IV arm. In the TCZ-SC–IV arm, oral glucocorticoid use was lower and RA duration was longer compared with other arms.
Efficacy
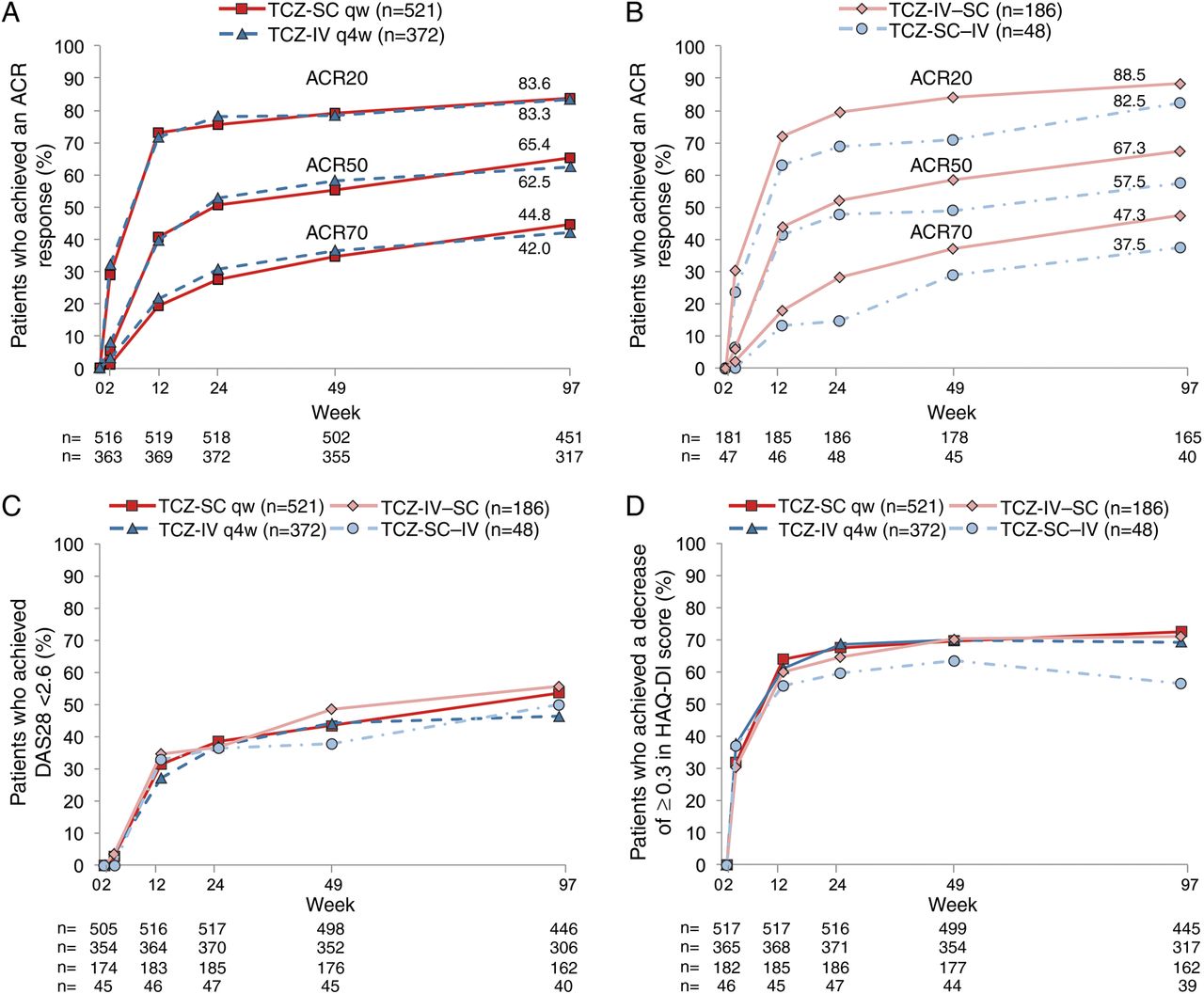
The proportions of completer patients who achieved an ACR20 response at week 97 were 83.6% in the TCZ-SC arm and 83.3% in TCZ-IV arm compared with 75.5% and 78.2%, respectively, at week 24 (figure 3A). In the TCZ-IV–SC and TCZ-SC–IV arms, ACR20 responses were maintained after switching (weeks 24, 97: TCZ-IV–SC 79.6%, 88.5%; TCZ-SC–IV 68.8%, 82.5%; figure 3B). ACR50/70 responses were similar to ACR20 responses across all arms. The ACR responses analysed in the ITT population showed similar results (see online supplementary figure S1).

Proportion of patients treated with either (A) subcutaneous tocilizumab (TCZ-SC; n=521) or intravenous tocilizumab (TCZ-IV; n=372) as well as (B) patients who switched from TCZ-SC to TCZ-IV (TCZ-SC–IV; n=48) and vice versa (TCZ-IV–SC; n=186) achieving 20%, 50% and 70% improvements per the American College of Rheumatology criteria (ACR20, ACR50 and ACR70), (C) remission based on Disease Activity Score using 28 joints (DAS28 <2.6) and (D) Health Assessment Questionnaire-Disease Index (HAQ-DI) response (HAQ-DI score decreases of ≥0.3) from baseline over 97 weeks (intent-to-treat population). The ns refer to completer analysis. For TCZ-SC and TCZ-IV, this refers to patients who continued on TCZ-SC or TCZ-IV from baseline through the open-label period; patients who switched are not included in this population. If a patient withdrew prior to week 97 but had efficacy measurements between weeks 85 and 96, the patient was still counted for efficacy analysis but not as a patient who completed week 97. qw, weekly; q4w, every four weeks.
The proportions of patients achieving DAS28 remission were similar in all groups at week 24 (≈37%) and were maintained to week 97 (TCZ-SC 53.4%, TCZ-IV 46.4%, TCZ-IV–SC 55.6%, TCZ-SC–IV 50.0%; figure 3C). Similar results were observed for DAS28 LDA (weeks 24, 97: TCZ-SC 54.5%, 67.0%; TCZ-IV 49.7%, 61.4%; TCZ-IV–SC 56.8%, 66.0%; TCZ-SC–IV 57.4%, 67.5%). Following the 24-week randomised controlled trials, 25 (5.0%) and 24 (6.8%) patients at weeks 49 and 11 (2.5%) and 7 (2.3%) at week 97 in the TCZ-SC and TCZ-IV arms, respectively, achieved their first DAS28 remission status.
The proportions of patients who achieved HAQ-DI response at week 97 were comparable between the TCZ-SC and TCZ-IV arms (TCZ-SC 72.4%; TCZ-IV 69.1%) and were sustained after switching (TCZ-IV–SC 71.0%; TCZ-SC–IV 56.4%; figure 3D). The mean HAQ-DI score improved from ≈1.6 at baseline (see online supplementary table S1) to ≈1.0 in all arms at week 24 and was maintained or decreased through week 97.
The proportions of patients achieving CDAI remission were maintained from week 24 through week 97 in the TCZ-SC and TCZ-IV arms (weeks 24, 97: TCZ-SC 13.5%, 27.2%; TCZ-IV 15.0%, 24.1%). Rates were similar at week 97 in patients who switched (TCZ-IV–SC 27.3%, TCZ-SC–IV 30.0%). Similar observations were noted for Boolean remission (see online supplementary figure S2).
In all body weight categories, the proportions of patients who achieved DAS28 remission were maintained from week 24 through week 97 (see online supplementary figure S3).
Pharmacokinetics and pharmacodynamics
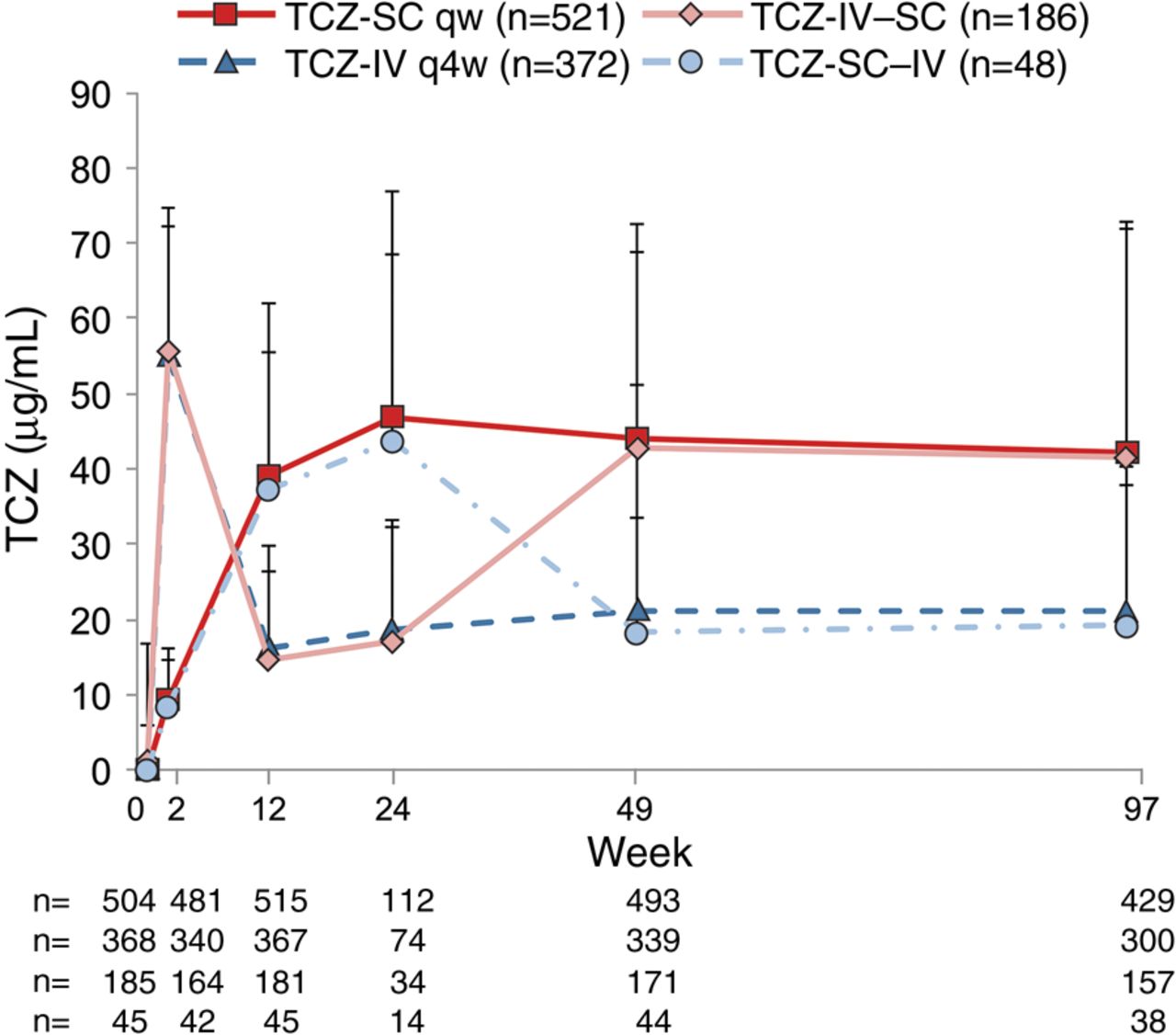
The mean TCZ concentrations in patients receiving TCZ-SC were approximately twice those in patients receiving TCZ-IV through week 97, consistent with observations through week 24. In patients who switched, concentrations were comparable to those in patients who received TCZ-SC or TCZ-IV continuously. In both switch arms, steady-state TCZ concentrations from week 25 onwards were not affected by the original dosing regimens (figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean (±SD) tocilizumab (TCZ) serum concentration over time (ITT-PK population) in patients treated with either subcutaneous TCZ (TCZ-SC; n=521) or intravenous TCZ (TCZ-IV; n=372) as well as patients who switched from TCZ-SC to TCZ-IV (TCZ-SC–IV; n=48) and vice versa (TCZ-IV–SC; n=186). ITT, intent-to-treat; PK, pharmacokinetic; qw, weekly; q4w, every four weeks.
Mean CRP concentrations decreased to below the upper limit of normal (ULN; 0.99 mg/dL) after the first dose of TCZ and remained within normal range through week 97 in all arms (see online supplementary figure S4). Mean ESR decreased during the first eight weeks in both TCZ-IV and TCZ-SC arms and remained low to week 97 in all arms (see online supplementary figure S5).
Safety
The safety profile of TCZ-SC remained stable over time. At week 97, AEs and serious AE (SAE) rates were comparable in the TCZ-SC and TCZ-IV arms, with the exception of injection site reactions (ISRs), which occurred more frequently in patients receiving TCZ-SC than in patients receiving TCZ-IV; however, the overall frequency of ISRs decreased over 97 weeks. All ISRs were Common Terminology Criteria for Adverse Events grade 1 or 2. The most common ISR symptom was erythema.
The safety profiles in patients who switched were similar to those continuing TCZ-SC or TCZ-IV (table 1 and see online supplementary table S2). Patient-year (PY)-adjusted AE rates in the TCZ-IV–SC arm were comparable to those in the TCZ-IV arm. Some AE rates were higher in the TCZ-IV–SC arm, but 95% CIs were wide and overlapped with the TCZ-IV arm; exceptions were hypersensitivity rates, which were lower, and ISR rate, which was higher than the TCZ-IV arm. The higher ISR rate after switching from TCZ-IV to TCZ-SC can be explained by multiple ISR episodes in five patients who reported ≥1 ISR symptom after almost every SC injection. The ISR events were generally non-serious (grade 1). Only one patient (TCZ-IV–SC arm) withdrew due to an ISR (erythema). None of the ISR symptoms in the TCZ-IV–SC arm were reported as SAEs. Although PYs in the TCZ-SC–IV arm were low (66.2 PY), PY-adjusted AE rates were overall comparable with those in the TCZ-SC arm.
Overview of cumulative AEs and serious AEs (SAEs) at week 97 (safety population*)
Infection rates decreased over time in all arms. Infections were the most commonly reported AEs (TCZ-SC 63.2%, TCZ-IV 57.8%, TCZ-IV–SC 17.7%, TCZ-SC–IV 56.3%) and led to withdrawal in 17 patients (2.7%) in the TCZ-SC arm, 12 (1.9%) in the TCZ-IV arm, 8 (4.3%) in the TCZ-IV–SC arm and 0 in the TCZ-SC–IV arm.
SAE rates were generally stable, consistent over 97 weeks, and comparable between the TCZ-SC and TCZ-IV arms. Serious infection rates were maintained over time. The rate of serious infections per 100 PY in the TCZ-IV–SC arm was higher (6.65 (95% CI 3.87 to 10.64)) than in the TCZ-IV arm (3.92 (95% CI 2.68 to 5.53)); however, the 95% CIs overlapped. The most common serious infections occurring in ≥2 patients in the TCZ-SC, TCZ-IV and TCZ-IV–SC arms were cellulitis and pneumonia. Only one serious infection (diverticulitis) was reported in the TCZ-SC–IV arm. Opportunistic infection rates were consistent over time and were atypical pneumonia (patient withdrawn), bronchopulmonary aspergillosis, oropharyngeal candidiasis and pharyngeal abscess in the TCZ-SC arm; genital herpes zoster and lepromatous leprosy (patient withdrawn) in the TCZ-IV arm; and genital herpes zoster, oropharyngeal candidiasis and Burkholderia pseudomallei infection (occurred in a major endemic region, Thailand) in the TCZ-IV–SC arm. Ten deaths were reported: four in the TCZ-SC arm (shock, cerebral infarction, thrombosis and unknown cause), four in the TCZ-IV arm (acute respiratory distress, cerebral infarction, sepsis and idiopathic pulmonary fibrosis) and two in the TCZ-IV–SC arm (pneumonia and sepsis).
No anaphylaxis cases were identified (according to Sampson criteria). Seven SAEs (five in the TCZ-SC arm and two in the TCZ-IV arm) were observed within 24 h of infusion/injection and evaluated as related to study treatment; of these, three were medically consistent with hypersensitivity and led to study withdrawal (two in the TCZ-SC arm and one in the TCZ-IV arm). One patient in the TCZ-SC arm and two in the TCZ-IV–SC arm experienced medically confirmed gastrointestinal perforations.
A similar frequency of elevated liver enzymes was observed in all treatment arms; there were no Hy's law cases. Most shifts in aminotransferase levels were from normal at baseline to ≤3× the ULN and occurred at a single time point only. The number of patients who experienced an alanine aminotransferase elevation ≥3× ULN in ≥2 consecutive samples over time was low. One patient experienced consecutive aspartate aminotransferase elevations ≥3× ULN (see online supplementary table S3). Most cases of neutropenia were newly occurring. The majority of events occurred at one or two consecutive visits (see online supplementary table S3). Also, 10 patients (1.9%) in the TCZ-SC arm, 10 (2.7%) in the TCZ-IV arm, 2 (1.1%) in the TCZ-IV–SC arm and 0 in the TCZ-SC–IV arm experienced grade 3 or 4 neutropenia on ≥2 consecutive visits. One patient (0.5%) in the TCZ-IV–SC arm had a serious infection of laryngitis (grade 3) that corresponded with grade 4 neutropenia. Neutropenia AEs leading to withdrawal remained low over time: four patients in the TCZ-SC arm and three in the TCZ-IV withdrew at week 97 versus one patient in each arm at week 24. No withdrawals due to neutropenia AEs occurred in either switch arm. Most patients maintained a platelet count above the lower limit of normal throughout treatment. One patient in the TCZ-IV arm experienced grade 3 thrombocytopenia (no grade 4; see online supplementary table S3). There was no association between grade 3 or 4 thrombocytopenia and serious bleeding events. The proportion of patients reporting shifts in total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol and triglycerides after initiation of TCZ was higher in the TCZ-SC arm than the TCZ-IV arm (total cholesterol 26.1% vs 21.0%; LDL 27.7% vs 23.2%; HDL 3.2% vs 3.9%; and triglycerides 23.8% vs 17.5%) at week 97, consistent with observations made at week 24 (see online supplementary table S3).
With the exception of the TCZ-SC–IV arm, patients weighing ≥100 kg at baseline had higher rates of AEs/100 PYs compared with patients in the lighter subgroups (see online supplementary table S4). The incidence of SAEs was comparable in the TCZ-SC and TCZ-IV arms across the lower body weight subgroups and higher in the TCZ-SC arm compared with the TCZ-IV arm in the ≥100 kg subgroup. The overall SAE rate in the TCZ-IV–SC arm was higher than in the TCZ-IV arm in each of the weight subgroups, whereas in the TCZ-SC–IV arm, it was lower than in the TCZ-SC arm.
Immunogenicity
The proportions of patients who developed anti-TCZ antibodies postbaseline were low: 10 patients (1.6%) in the TCZ-SC arm, 7 (1.1%) in the TCZ-IV arm, 0 in the TCZ-SC–IV arm and 1 (0.5%) in the TCZ-IV–SC arm. No correlation between anti-TCZ antibody development and clinical response or AEs was observed. Among patients who developed anti-TCZ antibodies, two in the TCZ-IV arm experienced ISRs. No patients with serious hypersensitivity events or who withdrew due to efficacy reasons developed anti-TCZ antibodies.
Discussion
Efficacy, safety and PK/PD profiles were maintained with continuous TCZ dosing through week 97. Efficacy in the TCZ-SC and TCZ-IV arms was comparable from weeks 24 to 97 for ACR20/50/70 response, DAS28 remission and HAQ-DI response rates. The efficacy and safety profile in patients who switched formulations was comparable to that in patients who remained on TCZ-IV or TCZ-SC. Lower ACR20/50/70 and HAQ-DI response rates occurred in the TCZ-SC–IV arm compared with the TCZ-SC arm; however, these lower values were observed from baseline through week 97. Differences may be due to the lower number of patients in the TCZ-SC–IV arm or to baseline differentiating factors in this group, such as shorter RA duration, higher number of failed aTNFs, lower rates of ACPA positivity and oral corticosteroid use. No new safety signals were identified, and the safety of TCZ-SC observed at week 24 was maintained with longer PYs. TCZ-SC had a safety profile similar to the known safety profile of TCZ-IV, with the exception of ISRs, which were more common with TCZ-SC.
In patients who switched formulations, the safety profile was comparable to patients who received the same formulation throughout the study. AE rates in the TCZ-SC–IV arm were comparable to those in patients who continued to receive TCZ-SC. In the TCZ-IV–SC arm, PY-adjusted AE rates during the OL period were generally comparable to those in the TCZ-IV arm and lower than in the TCZ-SC arm. SAE rates (including serious infections) were consistent over time up to 2 years. Serious infection rates were higher in the TCZ-IV–SC arm compared with the TCZ-IV arm. However, for both arms, SAE rates, including serious and opportunistic infections, had overlapping CIs, indicating no significant differences. Investigation into possible differences suggested that no baseline characteristics for age, weight, origin, prior aTNF treatment failure, baseline corticosteroid use or comorbidities appeared to favour the TCZ-IV arm. Comorbidities, such as diabetes, chronic obstructive pulmonary disease (COPD) and tobacco use, which predispose patients to infections, were comparable in the TCZ-IV–SC arm and the TCZ-IV arm. While there was a higher incidence of concurrent COPD in the TCZ-IV–SC arm (2.7%) compared with the TCZ-IV arm (1.8%), fewer patients in the TCZ-IV–SC arm (5.9%) had concurrent diabetes compared with the TCZ-IV (9.4%) and TCZ-SC arms (7.8%).
Immunogenicity of biological therapy has been a concern for healthcare providers. However, the proportions of patients who developed anti-TCZ antibodies remained low and were comparable up to week 97 in all treatment arms, including the switch arms. No correlation of anti-TCZ antibody development with clinical response or AEs was observed.
In all treatment groups and body weight categories, DAS28 response rates were maintained with dose continuation and after switching formulations. The data in the highest and lowest weight categories for the TCZ-SC–IV arm should be interpreted with caution due to the low number of patients in these groups. Infection risk factors, such as diabetes and COPD, could explain the numerically higher incidences of AEs in the TCZ-SC and TCZ-IV arms among patients weighing ≥100 kg at baseline compared with patients weighing <100 kg. A higher proportion of patients in the ≥100 kg category had diabetes or COPD (diabetes: TCZ-SC 22.6%, TCZ-IV 17.5%; COPD: TCZ-SC 4.8%, TCZ-IV 3.2%) compared with the 60 to 100 kg category (diabetes: TCZ-SC 6.6%, TCZ-IV 8.5%; COPD: TCZ-SC 2.8%, TCZ-IV 1.7%) and the <60 kg category (diabetes: TCZ-SC 4.9%, TCZ-IV 2.1%; COPD: TCZ-SC 0%, TCZ-IV 2.7%).
The long-term efficacy and safety of TCZ-SC was maintained and remained comparable with that of TCZ-IV, with the exception of ISRs. Efficacy in patients who switched between formulations was maintained, and safety profiles were similar to those in patients who remained on TCZ-IV or TCZ-SC. Most AE rates were comparable, with wide and overlapping 95% CIs. TCZ-SC could provide a more convenient administration option for patients with RA.
Acknowledgments
The authors wish to thank all investigators and patients who participated in the study and all members of the WA22762 study team.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Contributors MB and LR designed the study, analysed and interpreted the data. CD and GRB analysed and interpreted the data. GRB, AR-R, AC, SH, PL, DF, MJR, GR, CL, and EFM were involved in generating the data at their clinical research sites. All authors were involved in writing the manuscript and approved it.
Funding Support for third-party writing assistance for this manuscript, furnished by Denise Kenski, PhD, and Ramona Pufan, PhD, of Health Interactions, was provided by F. Hoffmann-La Roche, Ltd. Roche sponsored the study and participated in the design of the study, as well as in the collection, analysis and interpretation of the data. This manuscript was reviewed by Roche, but the decision to submit and publish this manuscript was contingent only upon the approval of the lead author and co-authors, including those employed by Roche.
Competing interests GRB has received research grants from Roche, Abbott, Pfizer, UCB, Merck Sharp & Dohme and Bristol-Myers Squibb; received consulting fees from Roche, Chugai, Pfizer, UCB and Bristol-Myers Squibb; and was on speaker bureaus for Roche, Pfizer, Merck Sharp & Dohme, Abbott and Bristol-Myers Squibb. AR-R has received research grants from Roche and Pfizer; received consulting fees from Roche, Chugai, Pfizer, UCB and Merck Sharp & Dohme; and was on speaker bureaus for Roche and UCB. AC has received research grants from UCB and Pfizer and received consulting fees from Roche, Chugai, Pfizer, UCB, Abbott and Bristol-Myers Squibb. PL has received consulting fees from Roche. MJR has received research grants from Roche. CL has received research grants from Roche, Bristol-Myers Squibb, Pfizer, Human Genome Science, Eli Lilly and Sanofi US and was on speaker bureaus for Bristol-Myers Squibb. EFM has received research grants and consulting fees from Roche and was on speaker bureaus for Roche. MB, LR and CD are employed by Roche.
Patient consent Obtained.
Ethics approval The study was conducted in accordance with the Declaration of Helsinki and good clinical practice.
Provenance and peer review Not commissioned; externally peer reviewed.