Article Text

Abstract
Objective To evaluate changes in structural damage and joint inflammation assessed by MRI following rituximab treatment in a Phase 3 study of patients with active rheumatoid arthritis (RA) despite methotrexate (MTX) who were naive to biological therapy.
Methods Patients were randomised to receive two infusions of placebo (n=63), rituximab 500 mg (n=62), or rituximab 1000 mg (n=60) intravenously on days 1 and 15. MRI scans and radiographs of the most inflamed hand and wrist were acquired at baseline, weeks 12 (MRI only), 24 and 52. The primary end point was the change in MRI erosion score from baseline at week 24.
Results Patients treated with rituximab demonstrated significantly less progression in the mean MRI erosion score compared with those treated with placebo at weeks 24 (0.47, 0.18 and 1.60, respectively, p=0.003 and p=0.001 for the two rituximab doses vs placebo) and 52 (−0.30, 0.11 and 3.02, respectively; p<0.001 and p<0.001). Cartilage loss at 52 weeks was significantly reduced in the rituximab group compared with the placebo group. Other secondary end points of synovitis and osteitis improved significantly with rituximab compared with placebo as early as 12 weeks and improved further at weeks 24 and 52.
Conclusions This study demonstrated that rituximab significantly reduced erosion and cartilage loss at week 24 and week 52 in MTX-inadequate responder patients with active RA, suggesting that MRI is a valuable tool for assessing inflammatory and structural damage in patients with established RA receiving rituximab.
Trial registration number NCT00578305
- Rheumatoid Arthritis
- Magnetic Resonance Imaging
- DMARDs (biologic)
- Inflammation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Inhibition of structural joint damage is a primary goal of rheumatoid arthritis (RA) therapy, and a number of disease-modifying antirheumatic drugs (DMARDs), including rituximab, have demonstrated this ability.1–5 While assessment of structural damage has traditionally been performed using radiography, the importance of earlier and more sensitive joint imaging has been recognised by recent guidelines.1 ,6 ,7
Although more expensive than radiography, MRI can simultaneously assess all relevant structures in the inflamed joint and is more sensitive at detecting bone erosions, allowing for smaller and shorter clinical trials.6 ,7 Furthermore, MRI visualises articular cartilage loss directly, whereas radiography evaluates cartilage loss indirectly through joint-space narrowing. MRI can also detect joint inflammation, which is particularly useful when assessing early or undifferentiated joint disease.8 ,9
Rituximab is approved in the European Union and the USA for the treatment of RA after inadequate response (IR) to ≥1 tumour necrosis factor (TNF) inhibitor. The approved dose of rituximab for the treatment of RA is two 1000-mg intravenous infusions separated by 2 weeks every 24 weeks or based on clinical evaluation, but not sooner than every 16 weeks. Rituximab inhibits joint damage progression in the indicated patient population, those with an IR to anti-TNF therapies (TNF-IR), and non-indicated populations.2 ,3 ,10
This study evaluated changes in structural damage and joint inflammation assessed by MRI following rituximab treatment in patients with active RA despite methotrexate (MTX) who were naive to biological therapy.
Methods
Study design and patients
RA-SCORE was a phase 3b, randomised, placebo-controlled, double-blind, multicentre, international study. Patients met the American College of Rheumatology (ACR) criteria for RA with a disease duration ≥3 months and ≤10 years, had active RA (Disease Activity Score in 28 joints (DAS28) C reactive protein score ≥3.2), and had experienced an IR to MTX (MTX-IR) at a dose of 12.5–25 mg/week for ≥12 weeks, with the last 4 weeks before baseline maintained at a stable dose. Minimal MTX doses of 7.5 mg/week or 10 mg/week were permitted only in cases of documented intolerance to higher doses. Patients were biological naive and were either positive for anticyclic citrullinated protein (≥20 U) or for rheumatoid factor (≥20 IU/mL). Patients were also required to have erosion and/or clinical signs and symptoms of synovitis in a single (MRI) joint (metacarpophalangeal and/or wrist). In patients with a disease duration of >1 year, clinical evidence of synovitis and ≥1 definitive radiographic erosion at screening based on central review were required. In those with disease duration of ≤1 year, clinical synovitis needed to be confirmed by MRI at baseline. Key exclusion criteria included a history of rheumatic autoimmune disease other than RA or significant systemic involvement secondary to RA. Secondary Sjögren's syndrome and secondary limited cutaneous vasculitis with RA were permitted.
Patients were randomised in a 1:1:1: ratio to receive two infusions of placebo, rituximab 500 mg, or rituximab 1000 mg intravenously on days 1 and 15. Premedication with analgesics, antihistamines and intravenous methylprednisolone 100 mg were required before each rituximab infusion. Patients continued to receive stable doses of MTX and folic acid/folate (≥5 mg/week). Concomitant oral glucocorticoids (at a stable dose ≤10 mg/day) were allowed; intra-articular glucocorticoid injections could be used in a limited fashion to treat severe RA flares. Rescue therapy, or increased dose of MTX or use of non-biological DMARDs, was permitted at week 16 for patients with <20% improvement in tender and swollen joint counts compared with baseline. Rituximab retreatment was permitted after week 24 if patients had a DAS28 C reactive protein score ≥2.6 and no contraindications to treatment (such as active infections).
This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Harmonised Tripartite Guideline for Good Clinical Practice (NCT00578305). The protocol and any accompanying material provided to the patient were approved by an Independent Ethics Committee or Institutional Review Board before starting the study. Patients provided written informed consent before enrolment.
Efficacy assessments
Patients returned for efficacy and safety assessments at weeks 4, 6, 12, 16, 24, 36, 44 and 52. MRI scans and radiographs were acquired at baseline (within 14 days before the first study medication infusion) and at weeks 12 (MRI only), 24 and 52. The most clinically inflamed hand and wrist (or the dominant hand, in case of equal inflammation) were imaged with a 1.5-Tesla whole-body MRI scanner using a commercial surface coil and an acrylic frame11 designed to ensure proper and reproducible positioning of the hand and wrist. The hand and wrist were scanned separately using coronal short-tau inversion recovery and coronal fat-suppressed three-dimensional gradient echo with and without intravenous gadolinium-based contrast agent. The same hand and wrist were used for all assessments.
All images were scored centrally by two independent radiologists who were blind to treatment allocations, clinical information and the order in which serial images were acquired. MRI images were scored using the Outcome Measures in Rheumatology Clinical Trials RA MRI Scoring (RAMRIS) method.6 ,12–14 Cartilage loss determined by MRI was assessed using a previously validated 9-point cartilage loss scale (CARLOS).11 ,15 Radiographs of hands and both feet were scored using the Genant-modified Sharp method.16
No progression was defined as a change from baseline in the RAMRIS erosion score of ≤0. Total damage scores were calculated using the following formula: RAMRIS erosion score+(2.5×CARLOS).15 Total inflammation scores were calculated using the following formula: RAMRIS osteitis score+(3×RAMRIS synovitis score).
Clinical efficacy parameters included ACR 20/50/70 and European League Against Rheumatism (EULAR) response rates and change from baseline in the DAS28-erythrocyte sedimentation rate (DAS28-ESR) and in the Health Assessment Questionnaire Disability Index (HAQ-DI). Safety parameters included treatment-emergent adverse events (AEs), clinical laboratory parameters, and human antichimeric antibody (HACA) to rituximab at week 24.
Statistical analyses
The intent-to-treat population was defined as all patients who were randomised and received any part of an infusion, with analysis groups defined according to randomisation. The safety population included all patients who received any part of an infusion, analysed according to the treatment received. Additional details are provided in online supplementary text 1.
In the primary analysis, the change in RAMRIS erosion score from baseline at week 24 (primary end point) was compared between patients receiving rituximab 1000 mg and those receiving placebo. Secondary end points included changes from baseline in the RAMRIS erosion score at weeks 12 and 52; RAMRIS synovitis and osteitis at weeks 12, 24 and 52; and proportions of patients with no progression in RAMRIS erosion score at weeks 24 and 52. Post hoc analyses evaluated for total inflammation scores at weeks 12, 24 and 52 and change in CARLOS and total damage scores at weeks 24 and 52.
The van Elteren test stratified for the factors used at randomisation (duration of MTX use < or ≥6 months and presence of bone erosions) was used for the primary analysis and to assess changes from baseline in RAMRIS erosion scores, Genant-modified Sharp radiographic erosion scores and CARLOS (imputation by linear extrapolation), and to assess changes from baseline in RAMRIS synovitis and osteitis scores (last observation carried forward). The Cochran-Mantel-Haenszel test was used to assess the proportions of patients with no newly eroded joints (no imputation used), of patients with no progression of erosions, of patients with improvements in synovitis and osteitis, and of patients achieving a EULAR and ACR 20/50/70 response (non-responder imputation). Analysis of covariance was used to assess changes from baseline in DAS28-ESR and HAQ-DI (last observation carried forward). All tests were two-sided and were conducted at the 5% significance level without correction for multiplicity.
Results
Patient disposition and baseline characteristics
A total of 283 patients from 37 centres in 17 countries were screened. Of these, 185 patients were randomised and received study drug and were therefore included in the intent-to-treat and safety populations (19 November 2007 to 12 April 2010 from first patient randomised to last patient visit). Of the patients receiving placebo, 22% failed to complete the study, compared with 5% and 7% of patients receiving rituximab 500 mg and rituximab 1000 mg, respectively. The predominant reasons for withdrawal in all treatment groups were non-safety related (see online supplementary table S1). Patient disposition is shown in online supplementary figure S1. The proportion of patients who remained in the study until week 52 was higher in the rituximab groups than in the placebo group.
Baseline demographics and disease characteristics were generally well balanced among the treatment groups (table 1). The mean age of the patients was 50 years, and the mean disease duration was slightly less than 5 years. At baseline, the mean DAS28-ESR was 6.2. Patients were receiving a mean MTX dose of >15 mg/week, and slightly more than half were receiving concomitant corticosteroids. Approximately 77% (48/62) and 72% (43/60) of patients in the rituximab 500-mg and rituximab 1000-mg groups, respectively, were retreated with a second cycle of rituximab after 24 weeks.
Patient demographics and characteristics (safety population) at baseline
MRI analysis
The analysis of the primary end point showed significantly less progression in the mean RAMRIS erosion score at week 24 in patients treated with rituximab 1000 mg compared with those treated with placebo (figure 1A). Similar results were observed at week 52. Significant differences were also observed between the rituximab 500-mg group and the placebo group at weeks 24 and 52. The mean change in RAMRIS erosion score at week 12 was not significantly different between either of the rituximab groups and the placebo group. The cumulative distribution plots revealed increased RAMRIS erosion score at week 24 in a greater proportion of patients in the placebo group than in either rituximab group (figure 1B).
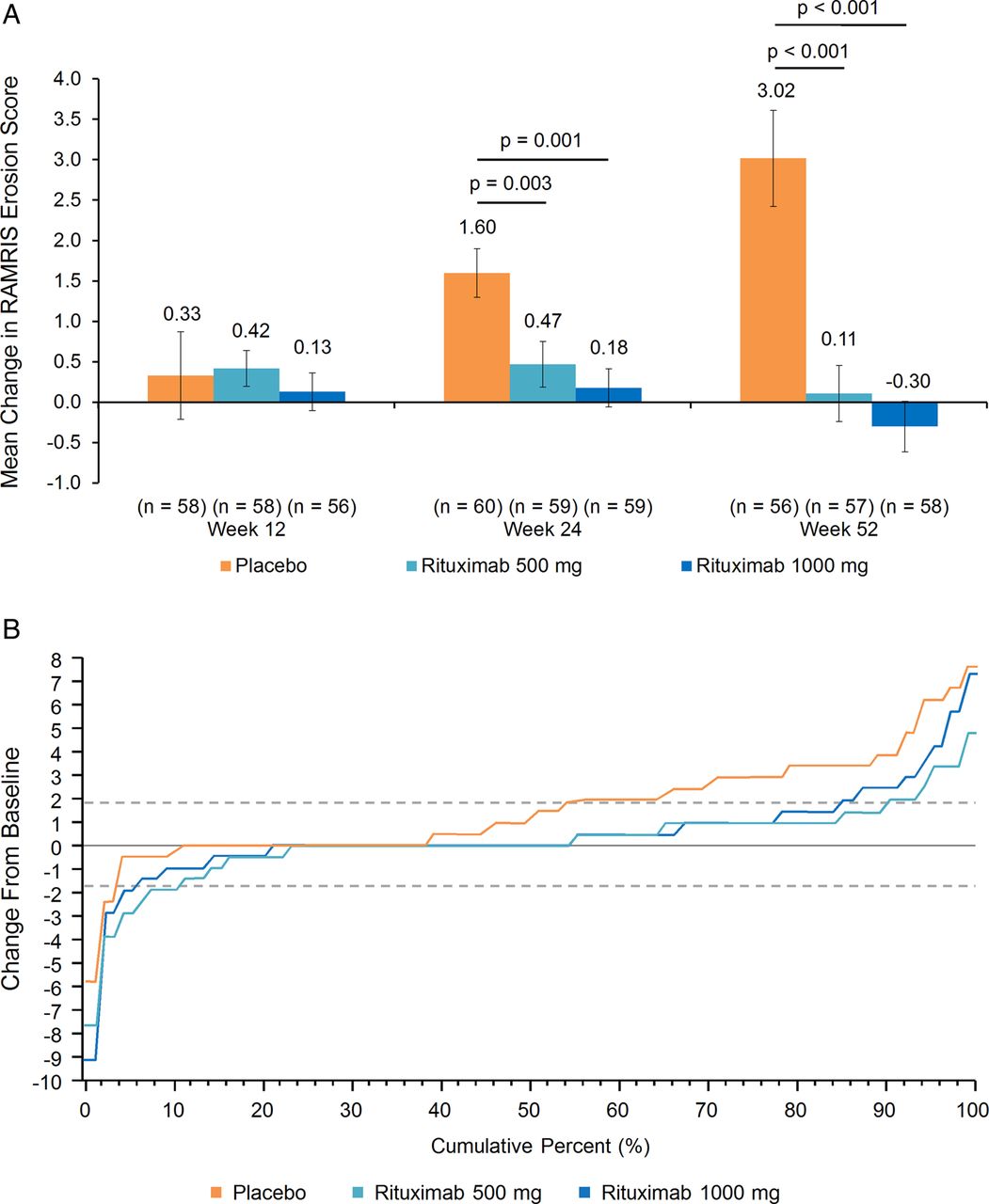
Primary end point. (A) Mean change in rheumatoid arthritis MRI Scoring (RAMRIS) erosion score (intent-to-treat (ITT) population). Missing values were imputed using linear extrapolation. Error bars represent the SE of the mean. (B) Cumulative distribution of change in RAMRIS erosion score at week 24. Missing values were imputed using linear extrapolation, ITT population. Broken horizontal lines represent ±SDC (smallest detectable change, determined according to the method of Bruynesteyn, et al.24 SDCs were 1.88 for RAMRIS erosion score, 1.54 for CARLOS, 2.11 for RAMRIS osteitis score and 1.56 for RAMRIS synovitis score).
Patients treated with rituximab 1000 mg demonstrated a progressively greater decrease in mean RAMRIS synovitis score at weeks 12, 24 and 52, which was significantly greater than placebo at each time point (figure 2A). Patients receiving rituximab 500 mg also showed a progressive decrease in the mean RAMRIS synovitis score at each time point; the decreases were numerically less than those in the rituximab 1000 mg-treated patients and were significantly different from those in the placebo-treated patients at weeks 24 and 52. The rituximab 1000-mg group demonstrated significantly greater reductions in RAMRIS osteitis scores compared with those in the placebo group at weeks 12, 24 and 52; similar results were observed in the rituximab 500-mg group at each time point (figure 2B). Mean cartilage loss scores based on CARLOS increased progressively at weeks 24 and 52 in patients treated with placebo, whereas patients treated with rituximab 1000 mg showed significantly greater improvement compared with those who received placebo (figure 2C) at week 52; a statistically significant difference was not observed at week 24. Patients receiving rituximab 500 mg demonstrated significantly less cartilage loss compared with those in the placebo group at weeks 24 and 52.

Mean changes in rheumatoid arthritis MRI Scoring (RAMRIS) synovitis, RAMRIS osteitis and MRI cartilage loss score (CARLOS) (intent-to-treat population). (A) Synovitis. Missing values were imputed using last observation carried forward. Error bars represent the SE of the mean. (B) Osteitis. Missing values were imputed using last observation carried forward. Error bars represent the SE of the mean. (C) Cartilage loss. Error bars represent the SE of the mean.
Patients who received rituximab 1000 mg demonstrated significantly better total damage scores compared with those in the placebo group at weeks 24 and 52 (figure 3A). Similar results were observed in the rituximab 500-mg group at weeks 24 and 52. Mean decreases in total inflammation score were significantly greater in the rituximab 1000-mg group compared with those in the placebo group as early as week 12 and were maintained through weeks 24 and 52 (figure 3B). Similar results were demonstrated in the rituximab 500-mg group.

{kind=link}
{kind=link}
{kind=link}
Mean change in total damage and total inflammation scores. (A) Total damage score. Total damage score=erosion score+(2.5×cartilage loss score). Missing values were imputed using linear extrapolation using baseline and week 12 images. Error bars represent the SE of the mean. (B) Total inflammation score. Total inflammation score=osteitis score+(3×synovitis score). Missing values were imputed using linear extrapolation using baseline and week 12 images. Error bars represent the SE of the mean.
The proportion of patients with no worsening of RAMRIS erosion score was significantly greater in the rituximab 1000-mg group than in the placebo group at week 24 (table 2) and week 52. Similar effects were observed for the rituximab 500-mg group, although the difference was only statistically significant at the 52-week time point.
Summary of efficacy outcomes
Radiographic analysis
At week 52, the analysis of the total radiographic Genant-modified Sharp score revealed significantly greater inhibition of radiographic damage in patients in the rituximab 1000-mg and 500-mg groups compared with the placebo group, although the study was not powered for the comparison of radiographic end points. The changes in mean erosion score from baseline were significantly lower in the two active treatment groups compared with that in the control group, whereas the changes in mean joint-space-narrowing score were numerically lower in both rituximab groups, but statistically significantly so in the rituximab 500-mg group
Clinical assessment
Clinical efficacy outcomes are presented in table 2. At weeks 24 and 52, patients who received rituximab 500 mg demonstrated significantly greater improvements in DAS28-ESR compared with those who received placebo; those who received 1000 mg demonstrated numerically greater improvements in DAS28-ESR scores compared with those who received placebo, but the difference was not statistically significant at either time point. Significantly more patients in both rituximab groups achieved ACR20 and ACR50 compared with patients in the placebo group at weeks 24 and 52. However, ACR70 rates at weeks 24 and 52 in either rituximab group were not consistently improved compared with the rate in the placebo group. At weeks 24 and 52, significantly more patients in the rituximab 1000-mg and 500-mg groups than in the placebo group had moderate or good EULAR responses. Significant improvements in HAQ-DI were seen in the rituximab 500-mg group at weeks 24 and 52. Numerical improvements were seen in the 1000-mg group compared with the placebo group at these time points, although these changes were not statistically significant.
Safety
The overall safety profiles were similar in both rituximab groups and are summarised in table 3. The incidence of AEs reported in the rituximab 1000-mg and 500-mg groups was numerically lower than that reported in the placebo group, as was the incidence of serious AEs per 100 patient-years. The incidence of infections was numerically higher in both rituximab groups compared with that in the placebo group, as was the incidence of serious infections per 100 patient-years. One patient in the rituximab 500-mg group reported a serious soft tissue infection (of suspected bacterial origin), which was treated and resolved after 11 days. Two serious infections were reported in the rituximab 1000-mg group: bronchitis and omphalitis due to Escherichia coli. The two infections resolved following treatment. One endometrial carcinoma was reported in the rituximab 1000-mg group. No deaths and no life-threatening AEs were reported. Additional safety data are provided in online supplementary tables S2 and S3.
Summary of safety events
The incidence of infusion-related reactions decreased from 0%, 4.8% and 15.0% in the placebo, rituximab 500-mg and rituximab 1000-mg groups during the first course of rituximab to 0%, 0% and 5.0% during the second course, respectively. The maximum Common Toxicity Criteria for Adverse Events grade of the 13 total events was moderate.
Overall, no safety concerns were raised by the laboratory data. At week 24, five patients in the rituximab 500-mg group (10.6%) and one in the rituximab 1000-mg group (3.8%) were positive for human HACA. At week 52, only one patient (2.8%) remained positive for HACA in the rituximab 500-mg group, whereas two patients (6.5%) remained positive for HACA in the rituximab 1000-mg group.
Discussion
In this placebo-controlled, double-blind, study, we used MRI to evaluate changes in structural damage and joint inflammation in MTX-IR patients with active RA who were biologicals-naive and received MTX plus either rituximab or placebo. The mean RAMRIS erosion score progressed in the placebo arm over 52 weeks at a rate of approximately 0.3 RAMRIS units per month, consistent with the rates observed in the placebo arms of other randomised controlled trials of RA using MRI.17 Cartilage loss also progressed at 24 and 52 weeks in patients treated with placebo. Compared with placebo, rituximab 1000 mg significantly reduced MRI erosion at 24 weeks (primary end point) and erosion and cartilage loss at 52 weeks. The rituximab 500-mg dose demonstrated improvements relative to placebo that were generally in the same range as observed with the higher rituximab dose, but the study was not powered to detect differences between the two doses in the observed range. The secondary end points of synovitis, osteitis, cartilage loss, total inflammation and total joint damage scores each improved significantly with rituximab compared with placebo as early as 12 weeks, and improved further at weeks 24 and 52. The proportions of patients with no joint damage progression, with improvements in synovitis and osteitis, and with moderate or good EULAR response were all significantly higher with rituximab than with placebo. For some disease activity indices, such as DAS28 and HAQ-DI, the rituximab 1000-mg dose demonstrated numerically positive trends, which were not always statistically significant.
Safety data were consistent with those previously reported for rituximab in biological-naive patients,3 ,18 ,19 and no new safety signals were detected. AEs were predominantly mild, and few severe or serious AEs and no deaths were observed with treatment. No major differences between the safety profiles of the 1000-mg and 500-mg doses were observed.
These results are consistent with the findings of previous studies that used radiography to demonstrate inhibition of joint damage progression by rituximab,3 ,5 ,20 and may be explained by the inhibitory effect of rituximab treatment on osteoclastogenesis.21 Recent EULAR and ACR recommendations recognise the superiority of MRI to clinical examination at detecting joint inflammation and recommend the use of MRI to predict response to treatment.1 ,7
This study had several strengths. It is the first published report of MRI's ability to discriminate suppression of cartilage loss in a multicentre randomised controlled trial in RA. This is important because exclusion of the assessment of cartilage loss in prior clinical trials has been an obstacle to accepting MRI as a substitute for radiography in clinical trials. Previous studies have demonstrated that articular cartilage loss is at least as important as bone erosion in determining long-term disability in patients with RA,22 and suppression of bone erosion does not always indicate that cartilage loss has also been suppressed.23 Inclusion of the assessment of cartilage loss in this study further allowed determination of total joint damage by MRI, analogous to the Total Sharp Score that is used in RA clinical trials to assess radiographic joint damage. A limitation of this study was that suppression of cartilage loss and radiographic outcomes were not measured at week 12, limiting the ability to compare cartilage loss against bone erosion or discriminate progression between the two imaging techniques. Additionally, although treatment effects in the rituximab 500-mg and 1000-mg groups largely overlapped, in a few of the end points (particularly cartilage loss and joint-space narrowing), one or the other dosing arm did not reach statistical significance compared with placebo. Because this study was not powered to compare the two active treatment arms, additional analyses were not performed to explore this apparent discrepancy. Of note, in the IMAGE study, only the approved 2×1000-mg rituximab dosing group exhibited significant inhibition of radiographic progression compared with the placebo group, whereas those in the rituximab 500-mg group did not—a treatment effect that was robust to several analyses.3 Lastly, it is important to highlight that patients in this study were biological naive and DMARD-IR; therefore, the results cannot be extrapolated to patients with more progressive or refractory disease, such as TNF-IR patients.
In conclusion, this study demonstrated that rituximab significantly reduced erosion and cartilage loss at weeks 24 and 52 in MTX-IR patients with active RA. These changes were preceded by reductions in synovitis and osteitis, and explain radiographic evidence that rituximab+MTX prevents joint damage in patients with active RA. Our results suggest that MRI is a valuable tool for assessing inflammatory and structural damage in patients with established RA receiving rituximab.
Acknowledgments
The authors thank all the investigators and patients involved in RA-SCORE.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Contributors CP conceived and designed the research, was involved in generating data at his clinical research site, assured data collection from the NHIH and analysed and interpreted the data. PE, PPT, MØ, KO, FNS and KP were involved in generating the data at their clinical research sites. JD contributed analysis tools and performed statistical analysis. CB analysed and interpreted the data and performed statistical analysis. LHG and AG analysed and interpreted the data. All authors were involved in writing the manuscript and approved it.
Funding This study was funded by Roche. Support for third-party writing assistance for this manuscript, furnished by Vivian Chen, PharmD, of Health Interactions based on the involvement and input of all authors, was provided by F Hoffmann-La Roche. All authors read and approved the final content of this manuscript.
Competing interests CP is a shareholder of Spire Sciences, Past Shareholder of Synarc (currently BioClinica), has undertaken clinical trials and/or provided expert advice for AbbVie, Acerta, Amgen, AstraZeneca, Bristol-Myers Squibb, Celgene, Eli Lilly and Company, Five Prime, Genentech, Janssen, Medimmune, Merck, Novartis, Pfizer, Rigel, Roche, Salix-Santarus, Sanofi, Samsung, UCB and Vertex, Employee of Spire Sciences, Past Employee of Synarc (currently BioClinica). PE has undertaken clinical trials and provided expert advice for Pfizer, MSD, Abbvie, Novartis, Roche and Bristol-Myers Squibb. PPT has served as a consultant for Roche/Genentech and became an employee of GlaxoSmithKline after completion of this work. JD is a consultant for Abbott, Amgen, AstraZeneca, Biogen-Idec, Bristol-Myers Squibb, BioClinica, Celgene, Centocor, Core Lab Partners, Crescendo, Eli Lilly and Company, Genentech, Genzyme, Merck, Icon Medical Imaging, Novartis, Perceptive Informatics, Pfizer, Rigel, Roche, UCB, VirtualScopics, Wyeth, Employee of Spire Sciences, and Past Employee of Synarc. FNS has received consulting fees from: Roche, Pfizer, UCB, Abbott and Meiji Seika, Speakers bureau fees from: BMS Roche, Pfizer, UCB, Abbott and Meiji Seika. KP has received consulting fees and lecturer/speaker fees from Roche, AbbVie, BMS, Pfizer, Amgen and MSD. M-AB, LHG, CB and AG are employees of F Hoffmann-La Roche.
Patient consent Obtained.
Ethics approval Independent Ethics Committee or Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.