Article Text

Abstract
Objectives The HIV restriction factor, SAMHD1 (SAM domain and HD domain-containing protein 1), is a triphosphohydrolase that degrades deoxyribonucleoside triphosphates (dNTPs). Mutations in SAMHD1 cause Aicardi–Goutières syndrome (AGS), an inflammatory disorder that shares phenotypic similarity with systemic lupus erythematosus, including activation of antiviral type 1 interferon (IFN). To further define the pathomechanisms underlying autoimmunity in AGS due to SAMHD1 mutations, we investigated the physiological properties of SAMHD1.
Methods Primary patient fibroblasts were examined for dNTP levels, proliferation, senescence, cell cycle progression and DNA damage. Genome-wide transcriptional profiles were generated by RNA sequencing. Interaction of SAMHD1 with cyclin A was assessed by coimmunoprecipitation and fluorescence cross-correlation spectroscopy. Cell cycle-dependent phosphorylation of SAMHD1 was examined in synchronised HeLa cells and using recombinant SAMHD1. SAMHD1 was knocked down by RNA interference.
Results We show that increased dNTP pools due to SAMHD1 deficiency cause genome instability in fibroblasts of patients with AGS. Constitutive DNA damage signalling is associated with cell cycle delay, cellular senescence, and upregulation of IFN-stimulated genes. SAMHD1 is phosphorylated by cyclin A/cyclin-dependent kinase 1 in a cell cycle-dependent manner, and its level fluctuates during the cell cycle, with the lowest levels observed in G1/S phase. Knockdown of SAMHD1 by RNA interference recapitulates activation of DNA damage signalling and type 1 IFN activation.
Conclusions SAMHD1 is required for genome integrity by maintaining balanced dNTP pools. dNTP imbalances due to SAMHD1 deficiency cause DNA damage, leading to intrinsic activation of IFN signalling. These findings establish a novel link between DNA damage signalling and innate immune activation in the pathogenesis of autoimmunity.
- Autoimmune Diseases
- Autoimmunity
- Fibroblasts
- Systemic Lupus Erythematosus
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
SAMHD1 (SAM domain and HD domain-containing protein 1) functions as a dGTP-dependent triphosphohydrolase which converts deoxyribonucleoside triphosphates (dNTPs) into the constituent deoxynucleoside and inorganic triphosphate.1 ,2 SAMHD1 was recently identified as the restriction factor that renders human myeloid cells non-permissive to HIV-1 infection by depleting the dNTP pool required for reverse transcription of the viral RNA genome.3 This is counteracted by Vpx, a primate lentivirus auxiliary protein that targets SAMHD1 for proteasomal degradation.4 ,5 Mutations of SAMHD1 cause Aicardi–Goutières syndrome (AGS), an infancy-onset inflammatory encephalopathy that phenotypically mimics congenital viral infection6 and shows overlap with systemic lupus erythematosus (SLE), a multifactorial disease characterised by autoimmunity against nucleic acids.7 Both disorders are characterised by the formation of antinuclear antibodies and constitutive activation of antiviral type 1 interferon (IFN).8 ,9 Indeed, type 1 IFN plays a central role in SLE pathogenesis by initiating a self-perpetuating feedback loop that drives autoantibody production.9
AGS is also caused by mutations of genes of the nucleic acid-metabolising enzymes, three prime repair exonuclease 1 (TREX1), ribonuclease H2 (RNASEH2A, RNASEH2B, RNASEH2C) and RNA-specific adenosine deaminase 1 (ADAR1).10–12 While these enzymes carry out diverse functions within the intracellular nucleic acid metabolism, their deficiency is thought to result in the accrual of endogenous nucleic acid species that are recognised as danger signals by sensors of the innate immune system, which trigger the pathogenic type 1 IFN response.13 These findings underline an important role of intracellular nucleic acid metabolism in the maintenance of immune tolerance by preventing an inadequate innate immune response induced by recognition of self nucleic acids.
Despite the antiretroviral property of SAMHD1, absence of viral infection is a cardinal feature of AGS, suggesting an intrinsic cause for activation of an innate immune response in patients with AGS. This notion is supported by the finding of a spontaneous cell-intrinsic antiviral response in SAMHD1-deficient mice.14 ,15 dNTPs are the building blocks of DNA synthesis during genome replication, and their intracellular concentration requires tight control to preserve genome integrity, as imbalances in DNA precursor pools can impede DNA replication/repair leading to DNA damage, cell cycle arrest or cell death.16 Here we provide evidence for an important role for SAMHD1 in genome stability, implicating DNA damage and/or the cellular response to DNA damage in the pathogenesis of inflammation and autoimmunity.
Methods
Human subjects
Primary fibroblast cell lines were derived from skin biopsy samples. The study protocol was approved by the ethics committee of the Medical Faculty, Technische Universität Dresden, and written informed consent was obtained from all patients or their parents.
Plasmids, transfection, RNA interference
Human SAMHD1 cDNA was cloned into pEGFP-C1 (green fluorescent protein (GFP)–SAMHD1) and pEGFP-N1 (SAMHD1–GFP) (Clontech). GFP was replaced by mCherry using AgeI and BsrGI. Human cyclin A cDNA was cloned into pEGFP-C1 (GFP–cyclin A). Cells were transfected with fluorescently tagged SAMHD1 or cyclin A as described.17 For RNAi, HeLa cells were transfected with Silencer Select small interfering (si)RNAs (Ambion, see online supplementary table S1) using Oligofectamine (Invitrogen).
Cell proliferation, cell cycle analysis and β-galactosidase staining
Human fibroblasts, HeLa and HEK293T cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 2 mM l-glutamine and antibiotics. In all experiments, passage-matched fibroblasts (passages 4–11) were used. Fibroblasts were seeded at a density of 2×105 cells/25 cm2 flask and counted at indicated time points using a Neubauer chamber. Cells were synchronised by serum starvation for 48 h and harvested at the indicated time points after cultivation in growth medium for propidium iodide staining or lysate preparation. Flow cytometry was performed on an LSR II (Becton Dickinson), and the data were analysed using FACSDiva and FlowJo. Cells were stained using the Senescence β-Galactosidase Staining Kit (Cell Signaling). The percentage of blue cells in eight randomly chosen visual fields per slide was determined by light microscopy.
Quantitative reverse transcription (RT)-PCR
RNA was extracted with the RNeasy Mini Kit (Qiagen) followed by DNase I digestion. Target gene expression (see online supplementary table S2) was determined by quantitative RT-PCR using Taqman Universal PCR Master Mix (Applied Biosystems) on an ABI7300 and normalised to GAPDH.
Intracellular dNTP quantification
dNTP levels were determined by a primer extension assay as described.18
In vitro phosphorylation assay
Recombinant human GST-SAMHD1 was generated and purified as described.18 SAMHD1 (15 ng) was incubated for 2 h at 30°C with recombinant cyclin A2/cyclin-dependent kinase (CDK)1, cyclin A2/CDK2 or cyclin E/CDK3 (Sigma-Aldrich) in the presence of 10 mM ATP according to the manufacturer’s protocol. Kinase reactions were subjected to immunoblotting with phospho-T592-SAMHD1 antibody and SAMHD1 antibody.
Statistical analysis
Comparison of means was carried out using Student’s t test. p<0.05 was considered significant. Data are presented as mean±SD or mean ± SEM as indicated.
Additional methods are described in supplementary methods.
Results
SAMHD1-deficient fibroblasts show impaired proliferation and cellular senescence
To investigate the consequences of SAMHD1 deficiency, we examined the cellular phenotype of primary fibroblasts from two patients with AGS both of whom presented signs of systemic autoimmunity including antinuclear antibodies, thrombocytopenia, cutaneous chilblain lesions and arthralgia.7 Patient 1 (AGS1) was compound heterozygous for R290H and Q548X, and patient 2 (AGS2) was homozygous for H167Y, which alters one of the two highly conserved histidine residues of the catalytic HD domain.7 We had previously shown that SAMHD1 protein is reduced or absent in AGS patient cells.17 Consistent with this, the intracellular levels of deoxyadenosine triphosphate (dATP) and deoxythymidine triphosphate (dTTP) were at least 15-fold higher than in wild-type fibroblasts (figure 1A). Patient cells proliferated significantly more slowly than wild-type cells (figure 1B). Flow cytometry of synchronised cells demonstrated that reduced proliferation was not due to increased apoptosis, as shown by the absence of a subG1 peak, but rather by a delay in cell cycle progression (figure 1C). This was accompanied by induction of the cell cycle inhibitors, p21 and p16, and an increase in β-galactosidase-positive cells, reflecting an increase in lysosomal activity consistent with cellular senescence (figure 1D,E). In addition, patient cells exhibited an enlarged and flattened morphology as well as senescence-associated heterochromatin foci within the nuclei, reflecting genome silencing (see online supplementary figure S1). These findings suggest that dysregulation of the intracellular dNTP pool due to SAMHD1 deficiency causes a cellular stress response leading to premature senescence.
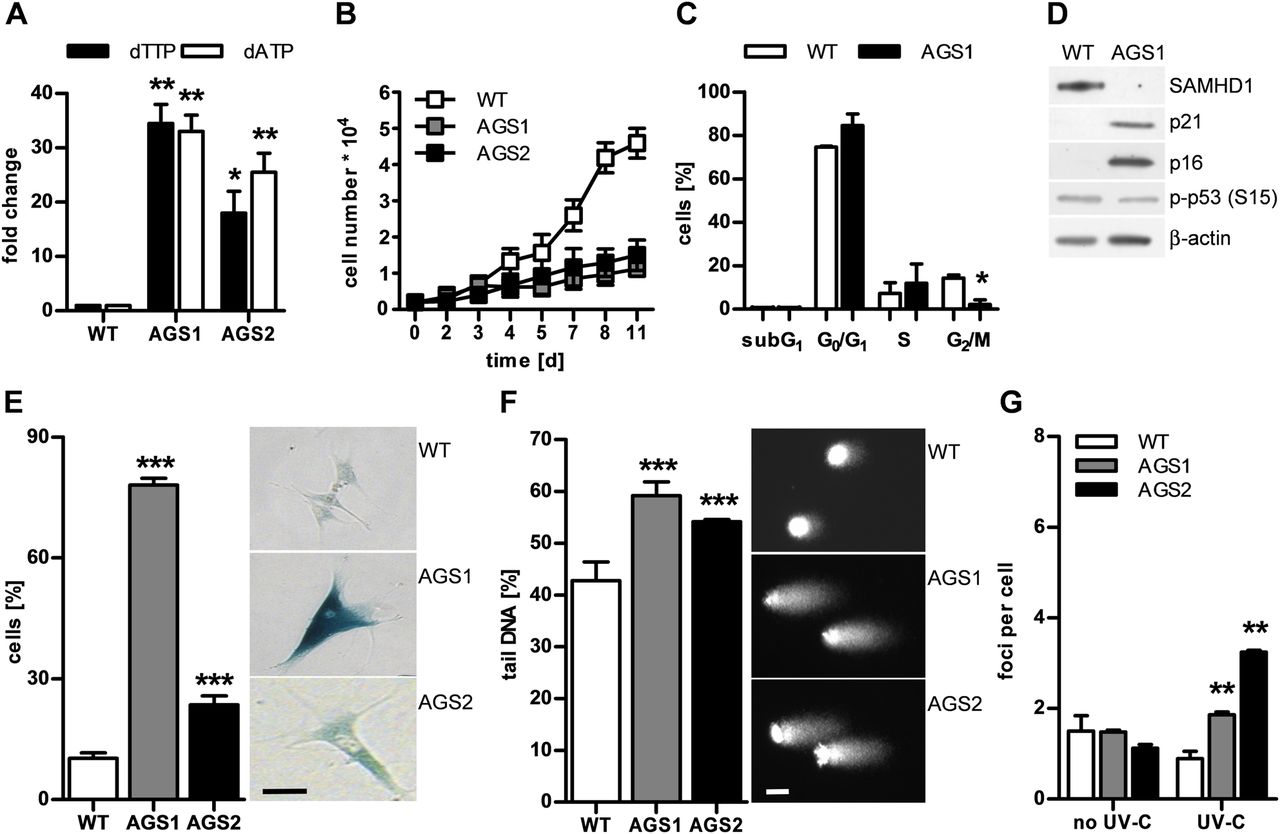
Dysregulation of deoxyribonucleoside triphosphate (dNTP) pools in SAMHD1-deficient fibroblasts causes genome instability leading to cell cycle delay and cellular senescence. (A, B) Fibroblasts from two patients with Aicardi–Goutières syndrome (AGS; AGS1, AGS2) exhibit markedly increased levels of deoxythymidine triphosphate (dTTP) and deoxyadenosine triphosphate (dATP) and proliferate more slowly than wild-type cells (WT, n=2). Shown are the means±SD of two independent experiments run in triplicate. (C) Synchronised AGS1 fibroblasts show delayed cell cycle progression, with an arrest in G1 and S as well as (D) induction of the cell cycle inhibitors, p21 and p16, but not of p53 phosphorylated at S15. (E) Patient cells exhibit a senescent phenotype shown by a large increase in β-galactosidase-positive cells. Scale bar, 100 μm. (F) Alkaline single-cell gel electrophoresis reveals global DNA damage as shown by increased comet tail length and irregular nuclear contour in patient fibroblasts compared with wild-type cells. Scale bar, 10 μm. (G) Native patient cells do not show increased DNA double-strand breaks as determined by counting of γH2AX- and 53BP1-double positive nuclear foci, but are more susceptible to DNA double-strand breaks in response to low-dose ultraviolet (UV)-C (20 J/m2). Values represent means±SEM of two (C, F, G) or three (D, E) independent experiments run in triplicate. *p<0.05, **p<0.01, ***p<0.001 vs wild-type by Student's t test.
DNA damage in SAMHD1-deficient cells is accompanied by type 1 IFN activation
Given the importance of balanced dNTP pools for genome stability, we examined SAMHD1-deficient cells for signs of DNA damage. We investigated the overall nuclear genome integrity by alkaline single-cell gel electrophoresis or comet assay, which detects alkali-labile sites such as single DNA strand breaks, abasic sites or incomplete excision repair sites, as well as stalled replication forks. In the absence of exogenous genotoxic stress, patient cells harboured significantly more DNA damage than wild-type cells, as indicated by formation of longer comet tails (figure 1F). Although native patient fibroblasts did not show more DNA double-strand breaks, as demonstrated by costaining of phosphorylated histone H2AX (γH2AX) and p53-binding protein 1 (53BP1), they were more sensitive to ultraviolet (UV)-C-induced DNA double-strand breaks (figure 1G, see online supplementary figure S2), indicating that pre-existing DNA damage renders SAMHD1-deficient cells more vulnerable to genotoxic stress. We further compared genome-wide transcriptional profiles of native patient fibroblasts with wild-type cells (figure 2A). In agreement with the cellular phenotype of SAMHD1-deficient fibroblasts, we observed upregulation of genes involved in DNA damage signalling such as CDKN1A (p21) and CDKN2A (p16) (figure 2B) and in senescence-associated metabolic changes (see online supplementary figure S3). Conversely, genes functioning in de novo and salvage pathways of dNTP synthesis and in cell cycle progression were strongly downregulated (see online supplementary figure S3). Interestingly, transcriptional profiling of patient fibroblasts also revealed upregulation of genes involved in immune activation including several IFN-stimulated genes (figure 2C), which was validated by quantitative RT-PCR (see online supplementary figure S4). These findings suggest that constitutive low-level DNA damage or chronic activation of DNA damage signalling can cause innate immune activation.

Genome-wide transcriptional profiles reveal upregulation of genes involved in DNA damage signalling and innate immune activation. (A) Heat maps based on RNA sequencing analysis represent hierarchically clustered transcripts showing up- or down-regulation by a factor of at least 2 in Aicardi–Goutières syndrome 1 (AGS1) and AGS2 compared with wild-type controls (WT, n=4). (B) Native patient fibroblasts exhibit constitutive upregulation of genes involved in the DNA damage response as well as (C) induction of interferon-stimulated genes. Indicated are the fold changes of the Reads Per Kilobase of exon per Million mapped reads (RPKM) values obtained by RNA sequencing analysis of patient fibroblasts (AGS1, AGS2) relative to the mean RPKM values of four WT cell lines.
SAMHD1 forms complexes with cyclin A in living cells
By affinity purification using GFP–SAMHD1 expressed in HEK293T cells as bait followed by MS analysis, we identified the cell cycle protein, cyclin A, as interactor. Cyclin A2 (GI:4502613) was confidently identified with two matching tryptic peptides: (R)EAGSALLALQQTALQEDQENINPEK and (R)AILVDWLVEVGEEYK (51 and 74 Mascot ion score, respectively). Interaction of SAMHD1 with cyclin A was validated by coimmunoprecipitation of cyclin A with GFP–SAMHD1 expressed in HEK293T cells. In addition, reverse coimmunoprecipitation of SAMHD1 using anti-cyclin A-coupled agarose beads confirmed interaction of SAMHD1 with cyclin A at the endogenous level (figure 3A, B). We also observed colocalisation of fluorescently tagged mCherry–SAMHD1 and GFP–cyclin A within the nucleus in HeLa cells (figure 3D). As SAMHD1 was shown to oligomerise,17 ,19 we addressed complex formation of SAMHD1 with cyclin A in living HeLa cells by fluorescence cross-correlation spectroscopy, a confocal microscope-based technique that allows assessment of mobility properties as well as interactions of fluorescently labelled molecules in vitro and in living cells.20 Cross-correlation between mCherry–SAMHD1 and GFP–cyclin A was 9.9±0.9% compared with 0.7±0.1% between monomeric GFP and mCherry, indicating formation of mobile complexes containing SAMHD1 and cyclin A within the nuclear environment (figure 3E). We further analysed the composition of these complexes by brightness analysis. The relative brightness of mCherry–SAMHD1 was on average twofold higher than monomeric mCherry, while that of GFP–cyclin A did not differ from monomeric GFP (figure 3E), suggesting that the formed complexes consisted on average of at least one cyclin A and two SAMHD1 molecules.

SAMHD1 is regulated by cyclin A in a cell cycle-specific manner. (A) Coimmunoprecipitation with N- or C-terminally green fluorescent protein (GFP)-tagged SAMHD1 expressed in HEK293T cells immobilised on GFP-Trap beads pulls down cyclin A and cyclin-dependent kinase 1 (CDK1), but not cyclin E. (B) Reverse coimmunoprecipitation with two different cyclin A antibodies (Ab 1, Ab 2) confirms interaction of SAMHD1 and cyclin A at the endogenous level. (C) In vitro kinase assay reveals phosphorylation of SAMHD1 at T592 by cyclin A/CDK1, but not by cyclin A/CDK2 or cyclin E/CDK3. (D) Cyclin A and SAMHD1 colocalise within the nucleus as shown by a high correlation of fluorescence signals in the scatter plot. Scale bar, 10 μm. (E) Fluorescence cross-correlation spectroscopy of living HeLa cells cotransfected with mCherry–SAMHD1 (SAMHD1) and GFP–cyclin A (cyclin A) shows a high cross-correlation, indicating formation of mobile SAMHD1–cyclin A complexes. Compared with monomeric GFP or mCherry, the average normalised brightness of GFP–cyclin A and mCherry–SAMHD1 is 1 or 2, respectively. At least 15 cells were measured per experiment. Data represent means±SEM from two independent experiments. ***p<0.001 by Student's t test. (F) HeLa cells synchronised at G0/G1 were lysed at the indicated time points after serum re-addition (SR) and immunoblotted with the indicated antibodies. (G) Intracellular deoxyribonucleoside triphosphate (dNTP) concentrations in HeLa cells treated as in (F).
SAMHD1 is regulated by cyclin A in a cell cycle-dependent manner
Consistent with our proteomic data, which also identified CDK1 as a SAMHD1 interactor, CDK1 was pulled down by GFP–SAMHD1 along with cyclin A (figure 3A). Cyclin A regulates cell cycle progression in association with cyclin-dependent kinases by binding and phosphorylation of numerous target proteins. Analysis of mass spectra for phosphorylated SAMHD1 peptides revealed phosphorylation of threonine at position 592 (T592, see online supplementary figure S5), which is part of a bona fide consensus sequence for known CDK substrates ((S/T*)PX(K/R), where S/T* is the phosphorylated serine or threonine and X any amino acid).21 We further examined recombinant SAMHD1 by in vitro kinase assay using an antibody to phospho-T592-SAMHD1 and observed phosphorylation of SAMHD1 by cyclin A/CDK1, but not by cyclin A/CDK2 or cyclin E/CDK3 (figure 3C), confirming that SAMHD1 is phosphorylated by cyclin A/CDK1.
Since cyclin A level is tightly controlled during the cell cycle, we hypothesised that it may regulate SAMHD1 expression in a cell cycle-dependent manner. To investigate this further, we examined SAMHD1 expression in HeLa cells synchronised at G0/G1 by serum starvation. Cell cycle phases were monitored by immunoblotting analysis of additional cyclins as cell cycle markers and by flow cytometry (figure 3F and data not shown). After release of synchronised cells into growth medium, SAMHD1 protein was almost undetectable during G1/S and gradually increased until late S phase (figure 3F). Corresponding to the course of SAMHD1 expression, dNTP levels were highest during G1/S, reflecting a high demand for DNA precursors during genome replication, and decreased continuously until G2/M (figure 3G). This was accompanied by the appearance of phosphorylated SAMHD1 paralleling cyclin A expression with a peak at G2/M transition. SAMHD1 then disappeared at the beginning of the subsequent cell cycle (figure 3F). These findings are in agreement with variable SAMHD1 expression and phosphorylation observed in resting and cycling human fibroblasts and monocytic THP-1 cells.22 ,23 Thus, SAMHD1 activity is subject to cell cycle-specific regulation at the post-translational level.
SAMHD1 knockdown recapitulates the senescent phenotype and IFN activation
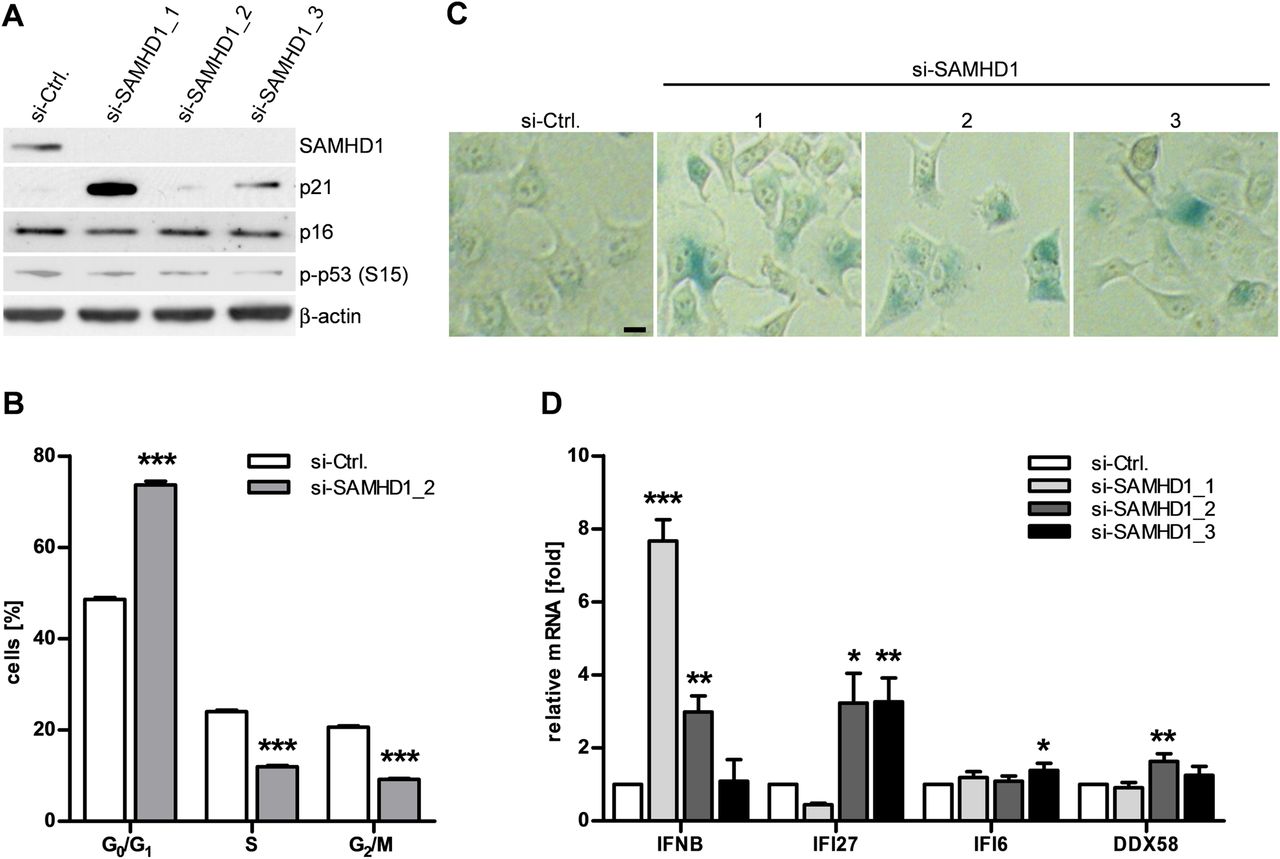
We next asked whether knockdown of SAMHD1 is sufficient to cause both activation of DNA damage signalling and type 1 IFN induction. Depletion of SAMHD1 in HeLa cells by RNA interference led to increased expression of p21 (figure 4A). This was accompanied by a delay in cell cycle progression and signs of cellular senescence, as shown by increased β-galactosidase activity (figure 4B,C). Furthermore, knockdown of SAMHD1 also led to transcriptional activation of IFNB and the IFN-stimulated genes, IFI27, IFI6 and DDX58 (figure 4D). Thus, depletion of SAMHD1 recapitulates the phenotypic changes observed in SAMHD1-deficient fibroblasts from patients with AGS.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
SAMHD1 knockdown recapitulates senescent phenotype and interferon (IFN) activation. (A) SAMHD1 is effectively knocked down in HeLa cells 72 h after transfection with SAMHD1-specific small interfering (si)RNAs (si-SAMHD1_1-3). Si-Ctrl, control-siRNA. This is accompanied by induction of p21. (B) Flow cytometry of propidium iodide-stained HeLa cells 72 h after transfection with si-SAMHD1_2 reveals delayed cell cycle progression and (C) an increase in β-galactosidase-positive cells. Scale bar, 10 μm. (D) SAMHD1 knockdown leads to induction of the IFNB gene and the interferon-stimulated genes IFI27, IFI6 and DDX58. Gene expression was normalised to GAPDH. Shown is the relative fold change in gene expression relative to control siRNA. Data are represented as mean±SEM from three (A) or four (B, C, D) independent experiments run in triplicate. *p<0.05; **p<0.01; ***p<0.001 by Student's t test.
Discussion
We show that SAMHD1-deficient fibroblasts from patients with AGS exhibit increased dNTP pools and chronic DNA damage. Constitutive activation of DNA damage signalling results in delayed cell cycle progression and cellular senescence. Senescence is a state of permanent cell cycle arrest and represents a cellular stress response that can be induced by various extrinsic and intrinsic stimuli including DNA damage.24 Remarkably, complete deficiency of the AGS-causing nucleases, RNase H2 and TREX1, in mice is also associated with activation of DNA damage signalling.25–27 This may suggest a common pathogenic mechanism in AGS involving DNA damage signalling. Furthermore, Trex1−/− mice exhibit increased lysosomal biogenesis,28 a cellular phenotype that is typically observed during senescence,24 raising the possibility that increased lysosomal activity may also contribute to AGS pathogenesis.
Transcriptional profiling of patient fibroblasts revealed induction of both genes of the DNA damage response as well as antiviral IFN-stimulated genes. Although upregulation of the IFNB gene was undetectable in patient fibroblasts, weak or intermittent type 1 IFN production cannot be excluded.29 Chronic low-level accumulation of intracellular nucleic acids may not be sufficient to induce type 1 IFN greatly. Alternatively, upregulation of IFN-stimulated genes, but not of IFNB, may reflect the complex transcriptional regulation of IFN gene expression with multiple positive and negative regulatory elements.30 Notably, the Irf3/STING/Tbk1-dependent activation of IFN-stimulated genes in Trex1−/− cells also occurs without detectable type 1 IFN induction.28 However, constitutive activation of antiviral genes such as STAT1 may prime IFN signalling and thereby enhance sensitivity to type 1 IFN stimuli.
dNTPs are the building blocks of DNA synthesis, and dNTP imbalances can have genotoxic consequences. Thus, changes in dNTP levels have been shown to impair replication fidelity, selection of replication origins, and fork progression.31–33 The DNA precursor pool is tightly regulated by an intricate enzyme network orchestrating dNTP synthesis and degradation to ensure replication of the genome with high fidelity once per cell cycle. Expression of ribonucleotide reductase, which converts ribonucleotides into deoxyribonucleotides during de novo dNTP synthesis, and of thymidine kinase 1, responsible for salvage of deoxynucleosides, varies during the cell cycle and is highest during S phase when DNA synthesis takes place.34 In agreement with previous reports,23 ,35 we identified cyclin A as an SAMHD1 interactor. Moreover, we demonstrate formation of mobile SAMHD1–cyclin A complexes in living cells consisting of at least one cyclin A and two SAMHD1 molecules, suggesting that binding of cyclin A may either require or promote oligomerisation of SAMHD1. As SAMHD1 represents a major negative regulator of DNA precursor pools in mammalian cells,22 our finding of cyclin A-dependent regulation of SAMHD1 provides a link between its dNTP-degrading activity and the cell cycle. Cyclin A is a key regulator of cell cycle progression and functions in association with cyclin-dependent kinases by phosphorylation of numerous target proteins. While association of cyclin A with CDK2 is required for passage into S phase, cyclin A also associates with CDK1 in AS phase, and this complex persists into late G2 phase, controlling the onset of mitosis.36 SAMHD1 is phosphorylated by cyclin A/CDK1 and fluctuates during the cell cycle, with the lowest levels observed in S phase when genome duplication takes place. A gradual increase in SAMHD1 during late S phase coincides with the appearance of phosphorylated SAMHD1 at the G2/M transition, which is followed by disappearance of SAMHD1. As phosphorylation of SAMHD1 by cyclin A/CDK1 does not interfere with catalytic activity,23 ,35 our findings may suggest that cyclin A/CDK1-mediated phosphorylation may be involved in degradation of SAMHD1 during the cell cycle. This would enable the cell to further adapt its dNTP supply to the changing physiological demands throughout the cell cycle. Given that the lentiviral factor, Vpx, can mediate proteasomal degradation of SAMHD1 in a DCAF1/E3 ligase-dependent manner,37 this raises the question of whether SAMHD1 may also be targeted for proteasomal degradation by as yet unknown physiological pathways.
The phenotype of AGS is characterised by inflammation and autoimmunity, which has been attributed to a type 1 IFN-mediated antiviral response induced by immune recognition of intracellular nucleic acids.13 Although cancer has not been described in children with AGS thus far, increased sensitivity to genotoxic stimuli in patient cells suggests that SAMHD1 may have tumour-suppressive properties. This is supported by the recent finding of somatic SAMHD1 mutations in patients with chronic lymphatic leukaemia.38 ,39 Patients with AGS are therefore predicted to have an increased cancer risk and should be closely monitored for malignant disease.
Our findings indicate that SAMHD1 maintains genome stability by regulating the DNA precursor pool, thereby preventing genotoxic dNTP imbalances. Loss of genome integrity in patient fibroblasts leads to upregulation of IFN-stimulated genes. Innate immune activation may be caused either by immune recognition of by-products of DNA repair or by as yet undefined signalling pathways of the DNA damage response. Since knockdown of SAMHD1 in HeLa cells is sufficient to recapitulate the phenotype of AGS patient fibroblasts, this supports a causal link between DNA damage signalling and innate immune activation. By limiting the dNTP supply available for DNA synthesis during replication and repair, SAMHD1 may also prevent aberrant synthesis of DNA species that could trigger a type 1-IFN-dependent immune response through activation of DNA sensors. Indeed, in Trex1−/− cells, type 1 IFN activation has been attributed to engagement of pattern-recognition receptors by cytosolic DNA, which may originate from DNA-repair processes or replication of endogenous retroviruses/retroelements.40 ,41 As SAMHD1 has been shown to restrict HIV by limiting the dNTP pool required for reverse transcription of its RNA genome,3 it may also restrict endogenous retroviruses/retroelements. This is supported by the recent finding that SAMHD1 can inhibit retrotransposition of a retroelement reporter in vitro.42 However, the exact origin of the nucleic acid species mediating innate immune activation in SAMHD1 deficiency remains to be investigated.
Collectively, these findings implicate chronic DNA damage as a cell-intrinsic source of an IFN-dependent innate immune response. The ensuing constitutive IFN signalling could eventually promote initiation of autoimmunity. These findings link pathways involved in DNA damage signalling with activation of a type 1-IFN-dependent immune response in the pathogenesis of autoimmunity.
Acknowledgments
We thank Susan Hunger and Kerstin Engel for excellent technical assistance, Nicole Berndt for help with γH2AX/53BP1 immunofluorescence, Andrea Knaust for assistance in MS analysis, and Patrick Keller (Max Planck-Institute of Molecular Cell Biology and Genetics, Dresden, Germany) for generation of antibody to phospho-T592-SAMHD1. We thank Axel Roers, Rayk Behrendt, Victoria Tüngler and Claudia Günther for helpful discussion. MAL-K, JG and AS are members of the Clinical Research Group 249, Medical Faculty, TU Dresden.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Handling editor Tore K Kvien
-
Correction notice This article has been corrected since it was published Online First. The penultimate sentence of the Methods section of the Abstract has been amended.
-
Contributors SK, CW, NK, WS, HL, LAN, BK, DA, AD and AS performed experiments and analysed and interpreted data. MH, AR, MCC and JG provided material and advice. SK, AS and MAL-K conceived experiments, interpreted data and wrote the manuscript.
-
Funding This work was supported by the Deutsche Forschungsgemeinschaft (LE 1074/4-1 to MAL-K, GU 612/2-2 to JG, SH 569/1-1 to AS), the Bundesministerium für Bildung und Forschung (02S8355 and 02NUK017D to MCC), a grant from the Friede Springer Stiftung to MAL-K, and National Institutes of Health Grants AI077401 and AI049781 to BK.
-
Competing interests None.
-
Ethics approval Ethics Committee, Medical Faculty, TU Dresden.
-
Provenance and peer review Not commissioned; externally peer reviewed.