Article Text

Abstract
Objectives Despite advances in systemic lupus erythematosus (SLE) treatment, many patients suffer from the disease and side effects. Atacicept is a fusion protein that blocks B-lymphocyte stimulator and a proliferation-inducing ligand, which are increased in patients with SLE.
Methods In this double-blind, placebo-controlled study, patients with moderate-to-severe SLE were randomised to atacicept 75 mg or atacicept 150 mg administered subcutaneously, or placebo twice-weekly for 4 weeks, then weekly for 48 weeks. Primary and secondary efficacy measures were the proportion of patients experiencing at least one flare of British Isles Lupus Assessment Group A or B, and time to first flare, respectively.
Results Enrolment in the atacicept 150 mg arm was discontinued prematurely due to two deaths. In the intention-to-treat population (n=461), there was no difference in flare rates or time to first flare between atacicept 75 mg and placebo. Analysis of patients treated with atacicept 150 mg suggested beneficial effect versus placebo in flare rates (OR: 0.48, p=0.002) and time to first flare (HR: 0.56, p=0.009). Both atacicept doses were associated with reductions in total Ig levels and anti-dsDNA antibodies, and increases in C3 and C4 levels. Most treatment-emergent adverse events were mild or moderate.
Conclusions There was no difference between atacicept 75 mg and placebo for flare rate or time to first flare. Analysis of atacicept 150 mg suggested benefit.
Trial registration number EudraCT: 2007-003698-13; NCT00624338.
- Autoantibodies
- B cells
- Autoimmunity
- Systemic Lupus Erythematosus
- Infections
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Morbidity and mortality have improved considerably for patients with systemic lupus erythematosus (SLE) in the past 50 years; the 4-year survival rate of 50% in 1950 is now a 10-year survival rate of 90%.1 However, some patients continue to die prematurely, suffer significant damage and have a reduced quality of life.1 A recent study2 of patients with lupus nephritis in a lupus cohort followed for 30 years found no change in the risk of the development of end-stage disease over this time and only a marginal improvement in mortality. The authors suggested that these results indicate that the benefits of conventional therapies (meaning steroids and immunosuppressives) have been maximised. The need to identify novel therapies with improved risk/benefit ratios remains, particularly for those responding inadequately to this approach. The use of biological drugs targeting key molecules or cells has yet to revolutionise the treatment of patients with SLE.
Several strategies have targeted B-lymphocytes due to their important role in autoantibody production, autoantigen presentation and immune dysregulation through cytokine secretion.3 The notion of directly attacking B-cells using rituximab (which binds the CD20 molecule on B-cell surfaces) seemed attractive, with numerous case series reporting success in ‘hard-to-treat’ SLE patients.4 However, subsequent controlled trials of rituximab were disappointing.5 ,6 An alternative approach to B-cell-directed therapy involves using a monoclonal antibody to a B-cell-activating factor known as B-lymphocyte stimulator (BLyS), a cytokine that promotes B-cell proliferation and differentiation. In two randomised, controlled Phase III trials,7 ,8 anti-BLyS (belimumab) plus standard-of-care (SOC) therapy demonstrated statistically significant benefit compared with SOC alone in patients with primarily cutaneous and musculoskeletal manifestations of SLE. Consequently, the drug was approved by the Food and Drug Administration and the European Medicines Agency.
Atacicept is a fusion protein containing the extracellular, ligand-binding portion of the receptor TACI (transmembrane activator and calcium-modulator and cyclophilin-ligand (CAML)-interactor) and the modified Fc portion of human IgG that blocks BLyS (like belimumab) and another B-cell activating factor, known as a proliferation-inducing ligand (APRIL).9 BLyS and APRIL levels are increased in patients with SLE,10 suggesting that dual blockade by atacicept may be more potent than blockading BLyS alone and has the benefit of targeting long-lived plasma cells in addition to B cells.11 Here, we report the results of a randomised Phase II/III trial of atacicept that sought to determine the efficacy and safety of atacicept in the prevention of flares in SLE.
Patients and methods
Study design
This was a 52-week, randomised, double-blind, placebo-controlled, parallel-group, multicentre trial (EudraCT: 2007-003698-13; NCT00624338) of atacicept in patients who originally had moderate-to-severe SLE exempting those with renal or central nervous system (CNS) disease. Informed consent, trial protocol and all substantial amendments were obtained from all patients in accordance with the relevant human patients’ Institutional Review Boards. The trial was conducted in accordance with the protocol, the International Conference on Harmonisation (ICH) guideline for Good Clinical Practice (GCP) and applicable local regulations as well as with the Declaration of Helsinki.
In order to evaluate better the effect of atacicept on the development of new flares, patients were administered prednisone or equivalent corticosteroid starting at 60 mg daily for 2 weeks (for patients with one or more British Isles Lupus Assessment Group (BILAG) A scores) and starting at 20 mg/day daily for patients with one or more BILAG B scores but no A scores. The steroid dosage was reduced from the start of week 3 in all patients (according to protocol) down to 7.5 mg daily at week 10. Patients achieving BILAG C or D scores in all systems at week 10 without any new A or B scores by week 12 (n=461) while on 7.5 mg of daily prednisone for weeks 11 and 12 were randomised to receive subcutaneous injection of atacicept 75 or 150 mg or matching placebo in a 1:1:1 ratio. Thus only patients with essentially inactive disease were eligible for randomisation. The dose of prednisone or equivalent remained constant during the study unless patients developed a postrandomisation flare. Flares were predefined as having an adjudicated BILAG A or B score due to items that were new or worse in any of the eight organ systems during treatment or imputed for subjects who had premature treatment discontinuation as defined in the statistical analysis plan. Randomisation was stratified by ‘black’, ‘white’ or ‘other’ and by BILAG category (A or B) at screening. Investigational medicinal drug was administered twice-weekly for 4 weeks and then once-weekly for the remaining 48 weeks. Patients were followed up for 24 weeks after the last dose of trial medication. A schematic diagram of the trial design as shown in the protocol is presented in online supplementary figure S1.
Medical history was obtained at study entry and a 4-weekly physical examination took place, with additional assessments at week 10 during the steroid reduction protocol and 2 weeks postrandomisation. Haematological and serum chemistry profiles were performed concomitantly and were evaluated using the Common Toxicity Criteria of the National Cancer Institute. Efficacy was assessed 4-weekly, primarily through a BILAG 2000 assessment 13.
The investigators were trained in the use of the BILAG assessment index and had to pass a written examination before enrolling patients. An adjudication committee regularly and carefully reviewed the patient’s assessments looking for inconsistencies that could be checked locally.
Inclusion and exclusion criteria
Patients aged ≥16 years (≥18 years old in Switzerland, Lithuania, Lebanon, Poland, Bulgaria, and site 120 in Australia) with active SLE defined by category A or B manifestations (excluding a single B score in haematology) on the BILAG index12 ,13 that required a change in steroid dose were included.
All patients had to have a diagnosis of SLE satisfying at least 4 of the 11 American College of Rheumatology classification criteria14 and updated in 1997,15 with a disease duration of ≥6 months. Patients had to be positive for antinuclear antibody (ANA) (HEp-2 ≥1:80) or anti-double-stranded DNA (dsDNA) antibodies (≥30 IU/L). Patients could be included if they had been on stable doses of azathioprine (≤3 mg/kg/day), hydroxychloroquine (≤400 mg/day), chloroquine (≤250 mg/day) or methotrexate (≤25 mg/week) for 2 months prior to screening (these patients remained on the same medication and dosage throughout the trial unless the drug was stopped due to toxicity).
Patients were excluded if they had taken any cyclophosphamide, mycophenolate mofetil (MMF), calcineurin inhibitors, leflunomide, 6-mercaptopurine or thalidomide within 3 months of screening. Other exclusion criteria included a history of treatment with rituximab, abatacept or belimumab, and participation in any interventional trial within the last 28 days or 5 half-lives (whichever was longer) from the trial. Patients with severe CNS lupus, congestive heart failure, a history of cancer, other than treated basal cell or squamous cell carcinoma of the skin or active moderate-to-severe glomerulonephritis (urinary protein:creatinine ratio >1.0 mg/mg and/or haematuria, or glomerular filtration rate <50 mL/min/1.73 m2), a history of recurrent or active infections such as HIV, tuberculosis, hepatitis B virus, or hepatitis C virus and a history of demyelinating disease, for example, multiple sclerosis or optic neuritis, were excluded.
Primary and secondary outcome measures
The primary outcome measure was the proportion of patients experiencing at least one flare of BILAG A or B, as defined by the BILAG index13 during the 52-week trial period. Premature discontinuation of study drug was imputed as a flare.
The main secondary endpoint was time to first flare during the treatment period. Other endpoints included the proportion of patients with a BILAG A or B flare within the first or second 24 weeks after randomisation, corticosteroid exposure postrandomisation, ordinal response categories for BILAG flares, changes from baseline in titres of anti-dsDNA antibodies and complement levels (C3 and C4).
The adjudication committee reviewed all BILAG data, including all renal and haematology results, whether attributed to lupus or not, but were blinded to immunology results (Ig, complement and B-cell levels), and treatment changes.
Pharmacodynamics
The pharmacodynamics of the study medication were assessed by measuring serum levels of immunoglobulins (total IgG, IgM, IgA), C3 and C4 complement and anti-dsDNA antibodies. Antinuclear (ANAs) and anti-dsDNA antibodies were measured using the ELISA test system.
Safety assessments
Safety was evaluated through the nature, incidence and severity of adverse events (AEs) and clinical laboratory abnormalities, and through assessment of any changes in clinical laboratory parameters, vital signs, ECGs and physical examination findings. Patients were tested for antibodies to atacicept at baseline and at the final poststudy visit 24 weeks after the last dose using bridging ELISA based on streptavidin precoated plates. Changes in antibody titres to pneumococcus toxoid, tetanus toxoid and diphtheria toxoid were evaluated. Titres of antibodies to pneumococcus, tetanus toxoid and diphtheria toxoid were measured at baseline and at week 52/early termination.
Statistical analyses
Data were analysed for the following populations: intention-to-treat (ITT): all randomised subjects; modified ITT (mITT): all randomised subjects who received at least one dose of study medication (this was also the Safety Analysis Set); and potential completer (PC): all subjects who were randomised ≥52 weeks prior to atacicept 150 mg arm termination. More details of the statistics used are provided in the accompanying supplementary materials.
Results
Study population
Of the 1007 patients enrolled, 546 failed screening, 461 were randomised (ITT analysis set) into three treatment groups and 455 of these received trial medication (mITT and safety analysis sets) (figure 1). Online supplementary table S1 summarises the reasons for screen failure, and online supplementary table S2 shows the reasons for discontinuation by treatment group during the treatment period.

Patient disposition. *Includes potential completer (PC) population, n=81; **Includes PC population, n=84; †Includes PC population, n=81.
Enrolment in the atacicept 150 mg arm was discontinued prematurely due to two deaths from pneumonias complicated by pulmonary haemorrhage. When treatment was discontinued, 62/144 patients in this arm had completed 52 weeks of treatment; 27 other patients had already been withdrawn for various reasons and, in the remaining 55 patients, treatment was stopped early as a safety precaution. Patients in the other two groups completed the protocol. The baseline characteristics between patients that completed the protocol and the PC population were similar. In total, 111 patients in the placebo group and 112 patients in the atacicept 75 mg group completed 52 weeks of treatment. Subjects who discontinued treatment prematurely, or who were randomised but not treated (three in the placebo group, two in the atacicept 75 mg group and one in the atacicept 150 mg group), were analysed as flared.
Demographic and baseline data were comparable across all treatment groups (table 1, see online supplementary table S3). More than 90% of patients in each treatment group were female. Approximately 70% in each treatment group were white, 18–21% were Asian and another 6–7% were classified as ‘other’. In the PC analysis set, 63–65% of patients in each treatment group were white, 21–26% were Asian and another 7–12% were classified as ‘other’. Less than 5% of patients in any treatment group were classified as black. Randomisation was stratified by disease severity and there was a similar number of patients with flares A or B across arms. The overwhelming number of patients with A or B flares were in the musculoskeletal or mucocutaneous systems.
Patient demographics and baseline disease characteristics
Efficacy
Primary endpoint
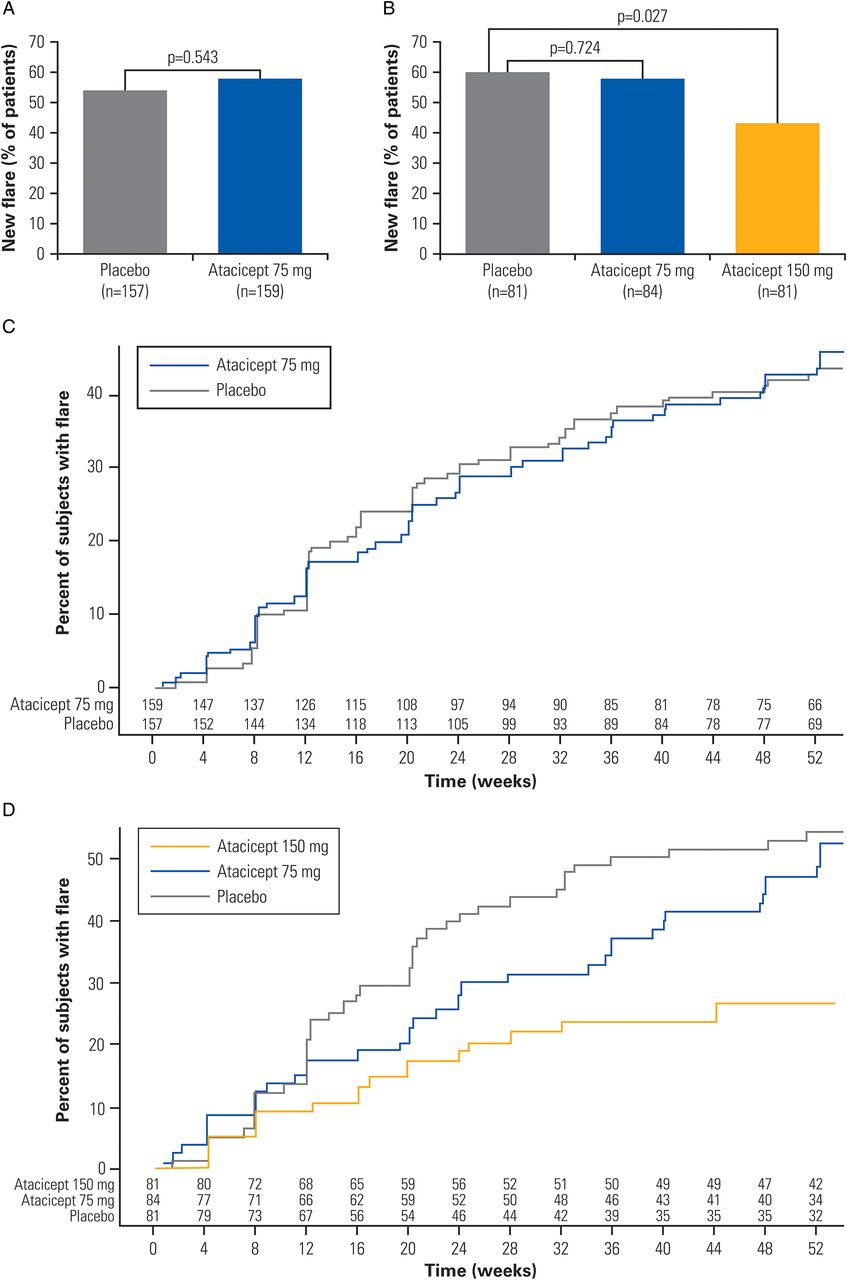
The primary objective was not met by the atacicept 75 mg group, which showed no benefit for atacicept 75 mg compared with the SOC plus placebo group in the ITT analysis set (flare rates of 58% and 54%, respectively; OR 1.15 (0.73 to 1.80), p=0.543) (figure 2A).

Analysis of the primary and the main secondary outcome measure. (A) Proportion of patients who experienced a flare during the 52-week treatment period in the ITT population; (B) proportion of patients who experienced a flare during the 52-week treatment period in the potential completer analysis; (C) time to first new flare in the ITT population; (D) time to first new flare in the potential completer population. ITT, intention-to-treat.
Post hoc analysis of atacicept 150 mg in the ITT analysis set suggested a beneficial effect of atacicept 150 mg versus placebo (flare rate 37% and 54%, respectively; OR 0.48 (0.30 to 0.77), p=0.002). Discontinuations due to arm termination were not imputed as flares. This analysis of the atacicept 150 mg group was undermined by the occurrence of two deaths in this group (see below). To address this confounding effect, we performed analyses of the primary endpoint in the PC population. PC population analysis paralleled the results of the ITT analysis. Specifically, there was no significant benefit of atacicept 75 mg compared with placebo (flare rate 58% and 60%, respectively; OR 0.89 (0.48 to 1.67), p=0.724). Atacicept 150 mg did show a beneficial effect versus placebo (flare rate 43% and 60%, respectively; OR 0.49 (0.26 to 0.92), p=0.027) (figure 2B).
Secondary endpoints
For the main secondary efficacy endpoint, time to first flare, there was no statistically significant difference between atacicept 75 mg and placebo for the ITT analysis set (HR 0.98 (0.69 to 1.40), p=0.929) (figure 2C). There was no significant difference in time to first flare in the placebo and atacicept 75 mg arms (HR (95% CI) 0.83 (0.53 to 1.29); p=0.404) for the PC analysis set (figure 2D).
The post hoc analysis showed atacicept 150 mg was associated with a significant delay in time to first flare for the ITT analysis set (HR 0.56 (0.36 to 0.87), p=0.009 (see online supplementary figure S2)). Patients in the PC population treated with an atacicept 150 mg dose had a 59% lower risk of having a new BILAG A or B flare over the 52-week treatment period than patients in the SOC plus placebo group (HR 0.41 (0.24 to 0.70), p=0.001, respectively) (figure 2D). Within the first 24 weeks of treatment, there was no significant difference across treatment groups for the proportions of flares (BILAG A or B). However, from weeks 24 to 52 of treatment, for those who completed the first 24-week treatment period, the atacicept 150 mg group showed a significant reduction in flare rate compared with the placebo group (16% vs 36% in the mITT analysis set and 15% vs 39% in the PC population). The mITT population was prespecified in the statistical analysis plan, but not in the protocol.
For ordinal response categories of BILAG flare, the odds of having a flare with lower severity (ordered from low to high: no A or B and completed treatment, no A and at least one B and at least one A (with or without B)) were higher for atacicept 75 mg (35 (48.6%), 29 (40.3%), 8 (11.1%); OR 1.20 (0.64 to 2.24), p=0.577) and significantly high for atacicept 150 mg (46 (70.8%), 16 (24.6%), 3 (4.6%); OR 3.24 (1.61 to 6.52), p<0.001) versus SOC and placebo in the PC population.
The reduction in the proportion of patients with a new flare as defined in the primary endpoint (at least one BILAG A or B) was observed in the eight systems evaluated in the atacicept 150 mg group as compared with atacicept 75 mg or placebo with a more marked decrease in the atacicept 150 mg group compared with the placebo and atacicept 75 mg groups in the musculoskeletal (18.1%, 21.7% and 9.0%) and mucocutaneous (17.5%, 21.0% and 9.0%) organ systems (placebo, atacicept 75 mg and atacicept 150 mg groups, respectively). Flares during the treatment period are summarised by organ system in online supplementary table S4.
A dose-dependent decrease was noted in the proportion of subjects in the PC analysis set who had at least one increase in corticosteroid dose to ≥20 mg/day. Thus, high dose of prednisone was given to 32%, 27% and 12% of patients in the placebo, atacicept 75 mg and atacicept 150 mg groups, respectively.
Pharmacodynamics
Analysis of total Ig levels revealed decreases in response to the corticosteroid treatment during the screening period followed by further atacicept treatment-associated decreases in a dose-dependent manner (figure 3). Over the 52-week treatment period, for treatment completers (all 285 subjects who completed 52 weeks of trial treatment), the median IgG concentration declined from baseline by 30% in the atacicept 75 mg arm and 38% in the atacicept 150 mg arm, compared with a 3% increase in the controls. Declines were also noted in IgA (53% and 58% decline, respectively, compared with a 2% increase in the controls) and IgM (66% and 69% decline, respectively, compared with a 1% decline in the controls). During screening according to the steroid reduction protocol, serum IgG showed a median percent decrease of approximately 8.5% across groups.

Change from baseline (median) in (A) IgG, (B) IgA and (C) IgM levels (treatment completer population). IgG, immunoglobulin G; IgA, immunoglobulin A; IgM, immunoglobulin M.
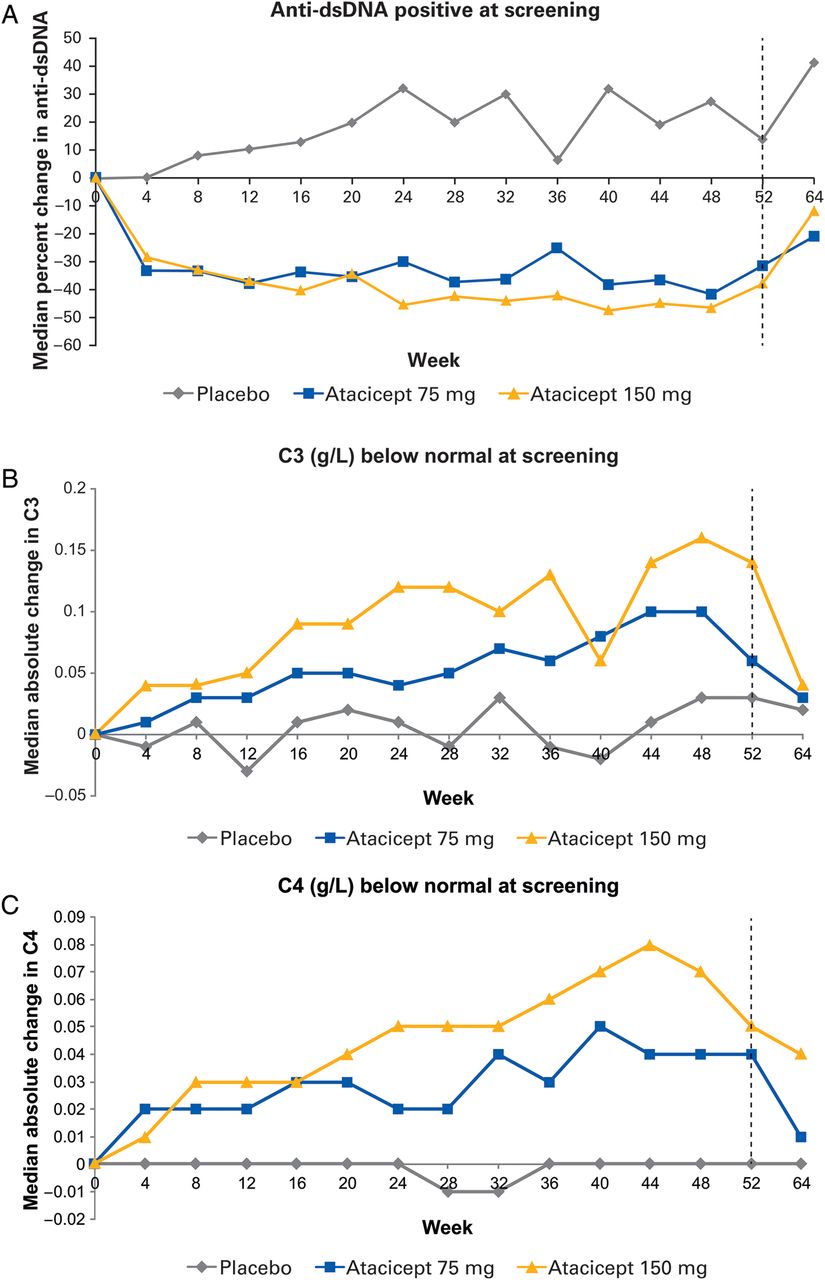
Treatment with atacicept also reduced the median concentration of anti-dsDNA antibodies (figure 4A). For those anti-dsDNA positive at screening (≥30 IU/mL), anti-dsDNA antibody levels were reduced from baseline by 31% in the 75 mg arm and 38% in the 150 mg arm versus a 14% increase in the control arm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Median change from baseline in anti-dsDNA and complement in the treatment completer population (all 285 subjects who completed 52 weeks of trial treatment). (A) Change in anti-dsDNA in patients positive at screening (≥30 IU/mL); (B) change in C3 in patients with below normal C3 levels at screening (<0.9 g/L); (C) change in C4 in patients with below normal C4 levels at screening (<0.1 g/L). Anti-dsDNA, anti-double-stranded DNA.
In both the ITT and the treatment completer analysis, atacicept was associated with increases in C3 levels at week 52 (least squares (LS) mean change vs placebo: 0.076 (p<0.001) and 0.138 (p<0.001) for atacicept 75 mg and atacicept 150 mg, respectively). This effect was more pronounced for patients with low levels at screening (<0.9 g/L). Thus, median changes in C3 from baseline to week 52 were 4.1%, 7.2% and 15.4% for placebo, atacicept 75 mg and atacicept 150 mg, respectively (figure 4B). Similar results were observed for C4 levels (figure 4C). LS mean changes versus placebo at week 52 were 0.046 and 0.066 for atacicept 75 mg and atacicept 150 mg (p<0.001 for both), respectively. In patients with low levels of C4 at screening (<0.1 g/L), median changes in C4 from baseline to week 52 were −0.4%, 42.7% and 49.5% for placebo, atacicept 75 mg and 150 mg, respectively. Following cessation of treatment, levels of total Ig, C3, C4 and anti-dsDNA returned towards baseline. There was also a reduction in B-cells and plasma cells.
Safety
Of 455 patients in the Safety Analysis set, 374 patients (82.2%) experienced one or more adverse events emerging during treatment (TEAEs) during the study (52-week treatment period and 24-week follow-up period), and 80 patients (17.6%) experienced at least one serious adverse event (SAE) during the treatment phase and/or follow-up phase (table 2).
Proportion of patients experiencing at least one adverse event (Safety Analysis Set)
The most commonly reported TEAEs were upper respiratory tract infection (17.1%), headache (15.6%) and urinary tract infection (14.1%). In general, incidence rates for patients in the placebo group tended to be lower than those for the atacicept 75 mg and atacicept 150 mg groups, with the exception of diarrhoea, sinusitis, influenza, hypertension and hypotension, which occurred in similar frequency. Of the 455 patients in the Safety Analysis set, most TEAEs were mild or moderate. The most frequently reported SAEs were infections and infestations, including pneumonia (10 patients: 2 patients in the placebo group, 5 patients in the atacicept 75 mg group and 3 patients in the atacicept 150 mg group). Five subjects receiving atacicept (two subjects receiving 75 mg, and three subjects receiving 150 mg) reported SAEs of pneumonia for which the events were assessed by the investigators as related to trial treatment. Two patients randomised to the atacicept 150 mg treatment group died. A 22-year-old male with SLE in the Philippines died from acute respiratory failure due to alveolar haemorrhage secondary to possible leptospirosis. This patient also suffered from an overlap syndrome with features of scleroderma, complicated by a history of recurrent infected digital ulcers. The second patient, a 30-year-old female in Argentina, died from pneumococcal pneumonia and alveolar haemorrhage secondary to lupus. These patients were treated with atacicept for 42 and 33 weeks, respectively, and were receiving prednisone 20 and 7.5 mg daily and hydroxychloroquine, respectively, for lupus at the time of the event. Both patients experienced reductions in their total IgG and IgM levels, but the IgG levels remained above 14.6 g/L (normal range approximately 6–18 g/L), and the IgM levels remained above 0.34 g/L. The trial's Independent Data Monitoring Committee (IDMC) recommended that the atacicept 150 mg dose group be discontinued.
Protective immunity to common pathogens was investigated. Vaccine titres against tetanus toxoid, diphtheria and pneumococcus were assessed at baseline and week 52 (or early termination). With regards to pneumococcal and tetanus titres, 1 in 30 (3.3%) placebo-treated patients had loss of protective titre status, whereas 1 in 10 (10%) atacicept-treated patients lost protective titre status. Median percent titre changes from baseline at week 52 for atacicept 75 mg, atacicept 150 mg and placebo, respectively, were −33.3%, −34% and 0 for tetanus toxoid; −21.1%, −11% and 0 for diphtheria; −29%, −30% and +2% for pneumococcus.
Immunogenicity was assessed by analysis of samples predose and postdose. Three patients in the atacicept 150 mg group showed measurable antibodies to atacicept at the week 24 follow-up visit.
Discussion
This study was designed to assess whether blockade of two B-cell activating factors, BLyS and APRIL, using atacicept could prevent flares in patients that had had lupus disease activity treated with (or that had responded to) a course of corticosteroids. Two fatal infections occurred in the 150 mg atacicept arm. A contributing role of atacicept could not be excluded, resulting in a premature termination of this group. Consequently, only the 75 mg and the placebo arm could be assessed for the primary endpoint analysis. There was no difference in the adjudicated flare rate between the placebo and the 75 mg atacicept arm.
Post hoc analysis of the primary endpoint in the ITT analysis set suggested a beneficial effect from the atacicept 150 mg regime since patients in this arm did show a statistically significant reduction the proportion of patients with BILAG flare A or B flare compared with placebo (36.6% vs 54.1%, OR 0.48 (0.30 to 0.77); p=0.002). The high-dose treatment was associated with a significantly delayed time to first flare (BILAG A or B) compared with placebo (HR 0.56, p=0.009). Among all patients who completed treatment prior to discontinuation of the 150 mg arm (81–84 patients per group), there was a distinct reduction in the flare rate and an increase in the time to flare for patients in the 150 mg arm compared with that seen in either the placebo or the 75 mg arm.
Atacicept may have steroid-sparing benefit. There was a dose-dependent decrease in the proportion of subjects who had at least one increase in corticosteroid dose to ≥20 mg/day. In spite of the early termination of the 150 mg arm, there remains a strong suggestion that the higher dose of atacicept was effective.
Serological improvements were seen in both atacicept arms. Both doses of atacicept reduced anti-dsDNA antibodies to a similar extent, so this does not explain the apparent clinical responses to the 150 but not 75 mg dose. The link between dsDNA antibodies and clinical activity is far from absolute.16 The beneficial effect on C3 (and to a lesser extent) C4 is more clear-cut in those treated with 150 mg atacicept, but why the beneficial clinical threshold appears at 150 vs 75 mg of atacicept remains incompletely understood.
The indication of possible efficacy must be weighed against the two deaths due to infection in the 150 mg arm. Thus, in the first belimumab trial7 in a similar range of non-renal clinical features patient population (Belimumab in Subjects with Systemic lupus erythematosus (BLISS)-52), there were nine deaths among 865 patients (1%), followed by three deaths among 819 patients (0.4%) in the BLISS-76 trial.8 In the phase IIb blisibimod trial in 547 patients with SLE, there were seven deaths, three in placebo and four among three active treatment arms.17
As both deaths in the current trial occurred in the 150 mg arm, the IDMC recommended discontinuation of treatment in this study arm as a cautionary measure. The infections in these two patients were not associated with hypogammaglobulinaemia. Treatment with atacicept has previously been found to reduce B-cells and Ig levels without significant adverse events in a dose-escalating Phase Ib study in mild/moderate non-renal lupus.9 A reduction in B-cells, plasma cells and the mean IgG concentration in the atacicept groups was observed in this study, with a 30% reduction in the 75 mg arm and a 38% reduction in the 150 mg arm, compared with a 3% increase in the placebo group. Median IgG levels in those receiving atacicept remained above the lower limit of normal over the 52-week treatment and only two patients reached the discontinuation threshold of 3 g/L. No serious infections were reported in these patients. The incidence of infection was comparable regardless of the degree of decline in IgG or IgM levels. In contrast, a trial of atacicept in combination with MMF and corticosteroids in patients with lupus nephritis was terminated prematurely due to rapid falls in IgG levels after six patients were recruited.18 This decrease in IgG started with the initiation of MMF and high-dose oral steroids 2 weeks before atacicept (four patients) or placebo (two patients) was given. Two of these atacicept-treated patients developed pneumonia, one due to Haemophilus influenza and one due to Legionella pneumophila before the trial was terminated, although neither patient died.
The overall similarity in adverse events between the three arms of the study, including serious events, is reassuring and consistent with previous studies of atacicept in rheumatoid arthritis, although the rate of total infections and serious infections was slightly higher with atacicept 150 mg, as observed in the rheumatoid arthritis19 and multiple sclerosis studies(unpublished data). The rate of serious infectious in the BLISS 76 study (7% and 7.3% in the 1 and 10 mg/kg arms, respectively) is virtually identical to the 6.9% observed in the atacicept 150 mg arm of the present study. In neither atacicept arm was the risk of serious infection statistically significantly increased compared with the placebo arm. In the Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial of rituximab in patients with non-renal SLE,20 the rate of serious infections was even higher (17% in the placebo group and 9.5% in the rituximab group), perhaps reflecting the more intensive use of glucocorticoids. These results suggest that contributing factors to the deaths observed in our study may have included the underlying disease, steroid therapy and delays in diagnosis and treatment.
The primary and main secondary efficacy endpoints in this trial were assessed using the BILAG assessment index. The flare rates seen in this study were in line with those previously reported.21 Distinct differences in flare rates and time to flare were seen between the treatment arms. Prevention of flares is an important clinically significant outcome as the frequency of flares is correlated with disease progression and organ damage.20
Atacicept is one of several B cell-directed therapies that have been under investigation in SLE, which include anti-CD20 (rituximab), anti-BLyS (belimumab, blisibimod and tabalumab), anti-CD22 (epratuzumab) and atacicept, which blocks BLyS and APRIL. In the current trial, we examined whether atacicept could prevent flares in patients at high risk for a lupus flare. We selected patients who had recently had a lupus flare that was controlled by a relatively short course of glucocorticoids. In contrast, the other therapies have been subjected to trial designs intended to treat active disease. Given the different trial strategies for each agent, and in the absence of any head-to-head comparison, it is not known which of these approaches will prove to have the most advantageous benefit: risk ratio or, alternatively, whether different agents will be appropriate for different purposes (eg, induction vs maintenance).
In conclusion, the primary endpoint of reducing the numbers of SLE patients who had a new flare was not met in the atacicept 75 mg arm compared with placebo. The results with the higher dose of 150 mg were more encouraging, but there are concerns about the possible infection risk in the light of two deaths in that arm. Treating patients with active lupus remains a great challenge.
Acknowledgments
The authors acknowledge Drs Michael Snaith and Asad Zoma who, with Prof Gordon, formed the Adjudication Committee; all investigators that participated in the trial and the atacicept study team; Stephen Wax, EMD Serono Inc, Rockland, ME, USA, for critical review of the manuscript; Gisèle von Büren, EMD Serono Inc, Billerica, MA, USA for publication coordination; and Medicus International, UK, for writing and editorial assistance, which was funded by EMD Serono, a subsidiary of Merck KGaA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Lay summary
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Tore K Kvien
Collaborators Principal investigators (125 sites): Argentina: Dr A Babini, Dr M Lazaro, Dr E Lucero, Dr C Merono, Dr O Rillo, Dr A Spindler, Dr G Tate, Dr B Velasco Zamora. Australia: Dr D Bossingham, Professor G Littlejohn, Dr D Nicholls, Professor G Reeves, Dr M Rischmueller. Austria: Dr O Zamani. Brazil: Dr E Sato, Dr M Scheinberg, Dr C Zerbini. Bulgaria: Professor R Rashkov. Croatia: Dr M Glasnovic, Dr D Martinovic-Kaliterna, Professor J Morovic-Vergles, Dr S Novak. Czech Republic: Dr D Tegzova, Dr P Vavrincova. France: Dr Z Amoura, Professor P Carle, Professor E Hachulla, Professor C Jorgensen, Professor J Sibilia, Professor J Viallard. Germany: Professor J Braun, Professor F Hiepe, Professor A Kuhn, Professor H Lorenz, Professor R Schmidt, Dr H Schulze-Koops, Dr J Wacker. Greece: Professor L Settas. India: Dr S Mouli Veeravalli. Israel: Dr A Balbir-Gurman, Professor P Langevitz, Professor D Mevorach, Dr Y Molad. Korea: Professor S Bae, Professor CS Cho, Professor HY Kim, Professor C Suh. Latvia: Professor D Andersone. Lebanon: Dr N Ziade. Lithuania: Assoc Professor A Baranauskaite, Dr S Stropuviene. Malaysia: Dr S Gun, Professor N Kong, Dr W Sulaiman. Mexico: Dr C Ramos Remus, Dr C Saldate Alonso. Netherlands: Dr T Huizinga, Dr P Tak, Dr P van Paassen. Philippines: Dr C Arroyo, Dr H Gomez, Dr E Perez, Dr E Ramiterre, Dr E Salido. Poland: Professor M Brzosko, Professor D Chudzik, Dr A Dyczek, Dr S Jeka, Professor S Sierakowski, Professor Z Zdrojewski. Russia: Professor O Barbarash, Professor O Ershova, Dr A Gordienko, Dr N Lomareva, Professor Y Shvarts, Professor V Sorotskaya, Dr N Vezikova, Professor S Yakushin. Serbia: Dr M Bogic, Professor A Dimic, Dr N Damjanov, Dr R Petrovic, Dr L Stojanovic. South Africa: Dr A Kalla, Dr I Louw, Dr S Nayiager, Professor H Reuter, Dr C Spargo, Dr D Whitelaw. Spain: Dr J Carbonell Abello, Dr A Fernandez Nebro, Dr J Gomez Reino, Dr F Lopez Longo. Switzerland: Professor J von Kempis. Taiwan: Dr CM Huang, Dr JL Lan, Dr HY Lin, Dr LB Liou. UK: Dr I Bruce, Dr H Tahir. Ukraine: Dr O Abrahamovych, Professor O Dyadyk, Professor Y Nikirenkov, Professor Y Rudyk, Professor S Smiyan, Professor M Stanislavchuk, Dr S Ter-Vartanyan. USA: Dr C Aranow, Dr A Askanase, Dr E Chakravarty, Dr W Chatham, Dr M Clark, Dr P Dhar, Dr A Dikranian, Dr J Fiechtner, Dr E Ginzler, Dr C Harmon, Dr W Knibbe, Dr E Lee, Dr M Lockshin, Dr M Luggen, Dr E Massarotti, Dr G Mitri, Dr S Peng, Dr N Singer.
Contributors DI was involved in the initial design of the study, designed data collection tools, taught and assessed potential collaborators, was involved in analysing the data, and drafted and revised the paper. CG was involved in setting up the methodology for lupus disease activity assessment in the trial, including BILAG assessment training, developed the Protocol for Adjudication of BILAG flares before the trial started, was Chair of the Adjudication Committee, was responsible for reporting on the adjudicated BILAG flares after reviewing all BILAG and relevant laboratory data at regular meetings and requesting any data clarification that was needed to enable the assessment of flares throughout the trial, provided advice on trial management as necessary, and was involved in data analysis and data interpretation as well as writing and revision of the manuscript, and approved the final version. DL contributed to the conduct of the study as Medical Director, and monitored data collection for the trial and data analysis. SC took part in the initial study design, data collection and performed the statistical analysis of the initial 52-week data. CPR contributed to the study design, conduct of the study as medical director, data collection, data analysis and writing of the manuscript. DW participated in developing the study design, interpreting the data and writing the manuscript from the first through the final draft.
Funding The APRIL-SLE study was funded by EMD Serono.
Competing interests DI reports other financial support from Merck Serono, outside the submitted work. CG reports personal fees from Merck Serono, during the conduct of the study; personal fees and non-financial support from UCB, other from Amgen, personal fees from BMS, Roche and GSK, outside the submitted work. CPR has a patent family: combination of BLyS and/or APRIL inhibition and immunosuppressants for treatment of autoimmune disease pending, and a patent family: dosing methods for treating autoimmune diseases using a TACI-Ig fusion protein such as atacicept, pending. DW reports personal fees from EMD Serono for assistance in study design, during the conduct of the study; personal fees from Genentech/Roche, Bristol-Myers Squibb and GlaxoSmithKline, outside the submitted work.
Ethics approval The trial protocol and all substantial amendments were approved by the relevant institutional review boards (IRBs) or independent ethics committees (IEs) and by health authorities, according to country-specific laws. A list of the IECs and IRBs responsible for oversight of the trial is presented in Appendix 16.1.3. This document can be supplied if requested.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Correction