Article Text

Abstract
Objective Previous work has suggested that the granulocyte macrophage colony stimulating factor (GM-CSF)–GM-CSF receptor α axis (GM-CSFRα) may provide a new therapeutic target for the treatment of rheumatoid arthritis (RA). Therefore, we investigated the cellular expression of GM-CSFRα in RA synovial tissue and investigated the effects of anti-GM-CSFRα antibody treatment in vitro and in vivo in a preclinical model of RA.
Methods We compared GM-CSFRα expression on macrophages positive for CD68 or CD163 on synovial biopsy samples from patients with RA or psoriatic arthritis (PsA) to disease controls. In addition, we studied the effects of CAM-3003, an anti-GM-CSFR antibody in a collagen induced arthritis model of RA in DBA/1 mice. The pharmacokinetic profile of CAM-3003 was studied in naïve CD1(ICR) mice (see online supplement) and used to interpret the results of the pharmacodynamic studies in BALB/c mice.
Results GM-CSFRα was expressed by CD68 positive and CD163 positive macrophages in the synovium, and there was a significant increase in GM-CSFRα positive cells in patients in patients with RA as well as patients with PsA compared with patients with osteoarthritis and healthy controls. In the collagen induced arthritis model there was a dose dependent reduction of clinical arthritis scores and the number of F4/80 positive macrophages in the inflamed synovium after CAM-3003 treatment. In BALB/c mice CAM-3003 inhibited recombinant GM-CSF mediated margination of peripheral blood monocytes and neutrophils.
Conclusions The findings support the ongoing development of therapies aimed at interfering with GM-CSF or its receptor in various forms of arthritis, such as RA and PsA.
- Rheumatoid Arthritis
- Treatment
- Pharmacokinetics
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease of the synovial joints and is associated with considerable morbidity, disability and mortality. It affects approximately 1% of the population. Although its aetiology is unknown, our understanding of the cellular and molecular mechanisms that contribute to the pathogenesis in RA has allowed the development of new successful treatments for RA. However, there is an ongoing need for new therapeutics targeting different molecules, because a substantial number of patients remain with active disease.
Several lines of data suggest that granulocyte macrophage colony stimulating factor (GM-CSF) and macrophages strongly influence the development and pathogenesis of RA as reviewed recently.1–4 GM-CSF is a cytokine initially defined by its ability to promote the formation of myeloid cells from bone marrow precursor cells. In addition, GM-CSF mediates the functional activation of mature neutrophils, eosinophils and macrophages—and therefore is associated with a number of inflammatory disorders such as RA.5 Clinical support for a link between GM-CSF and inflammatory arthritis has been suggested through a number of case reports that describe exacerbation of established inflammatory disorders including RA following treatment with exogenous GM-CSF.6–8
GM-CSF functions through a high-affinity heterodimeric receptor composed of a GM-CSF receptor specific α-subunit (GM-CSFRα) and a signal-transducing subunit, the common β-chain (βc) that is shared with the receptors for interleukin 3 (IL-3) and IL-5.5 ,9 GM-CSF and its receptor are found in the plasma, synovial fluid and synovial tissue (ST) of patients with RA.10–12 Early phase clinical trials have suggested beneficial effects of GM-CSF or GM-CSFRα inhibition in patients with RA.13–15 Taken together, these data support the involvement of GM-CSF and its receptor in the pathogenesis of RA. To further explore the potential of targeting this axis as a novel therapy for RA we carefully investigated the cellular expression of GM-CSFRα on macrophages in RA ST compared with disease controls and studied the effects of GM-CSFRα inhibition in vitro as well as in an animal model of RA. The results strongly support the development of anti-GM-CSFRα antibody treatment as a new therapeutic strategy in RA.
Methods
Patients
We obtained ST from 26 patients with RA classified according to the 1987 American College of Rheumatology criteria16 and from 24 patients with psoriatic arthritis (PsA) according to the Classification Criteria for Psoriatic Arthritis17 by mini-arthroscopy as described previously.18 All patients with RA and PsA had active disease defined by a disease activity score evaluated in 28 joints >3.2. We included ST of 9 healthy controls (HCs) and 10 patients with osteoarthritis (OA) undergoing exploratory arthroscopic surgery of a knee or ankle as controls. We acquired written informed consent of all participants prior to study participation as approved by the Medical Ethical Committee of the Academic Medical Center/University of Amsterdam.
Immunohistochemistry
We fixed and stained frozen serial sections with mouse monoclonal antibodies to GM-CSFRα (clone 4H1; Lifespan BioSciences, Seattle, Washington, USA) and CD68 (clone EBM11; Dako, Glostrup, Denmark) to detect macrophages. GM-CSFRα was stained using a 2-step immunoperoxidase method and CD68 was stained using a 3-step immunoperoxidase method as described previously.19 Control sections were stained with isotype control matched primary antibodies. Stained sections were analysed by computer-assisted image analysis for all markers using the QWin image analysis system (Leica, Cambridge, UK) as previously described.20 The number of positive cells was measured as stained cell count/mm2.
Immunofluorescence
To investigate GM-CSFRα expression by macrophages we incubated synovial biopsies of 5 patients with RA overnight at 4°C using primary antibodies specific for mouse antihuman GM-CSFRα (clone 4H1; Lifespan BioSciences), followed by Alexa Fluor 488 goat antimouse IgG2b antibody (Invitrogen, Carlsbad, California, USA). Subsequently, mouse monoclonal antibodies specific for macrophages (CD68; clone Y1/82A and CD163; clone GHI/61; Biolegend, San Diego, California, USA) were incubated for 60 min at room temperature and were detected using Alexa Fluor 633 goat antimouse IgG1 antibody (Invitrogen). We used a Zeiss LSM 780 Zen confocal microscope (Jena, Germany) to visualise staining.
Pharmacokinetic analysis of the mouse anti-GM-CSFRα antibody CAM-3003
We generated an antimouse GM-CSFR neutralising antibody (CAM-3003) (see online supplementary methods) and determined its pharmacokinetic profile in a single ascending dose study in female 8-week-old CD1(ICR) mice of approximately 20–22 g (see online supplementary table S1).
In vitro granulocyte CD11b expression assay
As cellular trafficking and adhesion has been identified as a key mechanism to recruit and retain inflammatory cells within the arthritic joint1 ,21 ,22 we explored whether GM-CSFRα inhibition impacts on the expression of the integrin CD11b. Bone marrow was obtained from femurs of approximately 8-week-old BALB/c mice (Charles River Laboratories, Margate, UK). Cells were plated in media (RPMI (Gibco, Paisley, UK)+1% v/v penicillin/streptomycin (Gibco) at 5E05/well in 96 well plates (Greiner, Frickenhausen, Germany)). First, CAM-3003 (Medimmune, Cambridge, UK) or isotype (R&D systems, Minneapolis, Minnesota, USA) was serial diluted in media and preincubated with cells for 30 min at 37°C. Murine GM-CSF (R&D systems) was added at a final assay concentration of 2.5 ng/mL. Second, recombinant GM-CSF was serial diluted from 100 ng/mL in media and cells were incubated with GM-CSF at 37°C for 1 h, washed with flow assisted cell sorting (FACS) buffer (2% v/v BSA (Sigma, St. Louis, Missouri, USA), 2% foetal calf serum (FCS) (Gibco), 2 mM EDTA (Sigma)) at 4°C. Anti-CD16/CD32 monoclonal antibody (MAb) (BD Pharmingen, San Jose, California, USA) was added at 0.5 µg/well as a Fc block and incubated at 4°C for 30 min and 0.1 µg/well of antimouse-CD11b PE Cy7 (eBioscience, San Diego, California, USA) and 0.125 µg of antimouse-Ly6G (GR-1) fluorescein isothiocyanate (FITC) (eBioscience) were incubated with cells at 4°C for 1 h. Cells were washed in FACS buffer and fixed in 2% v/v formaldehyde in phosphate buffered saline (PBS) and analysed using FACS Canto II flow cytometer (BD Biosciences, San Jose, California, USA). Data were processed using FlowJo software (Tree Star, Ashland, Oregon, USA) and expressed as geometric mean.
In vivo margination assay
On Day 0 female BALB/c mice (n=7–8/group) were injected intraperitoneally with a dose response of CAM-3003 (10 mg/kg, 1 mg/kg and 0.1 mg/kg), CAT004 (isotype control 10 mg/kg) or vehicle alone. Twenty-four hours post dosing mice were injected subcutaneously with mouse GM-CSF (0.25 μg; ProSpec Tany TechnoGene Limited, Rehovot, Israel) and peripheral blood collected by cardiac puncture following terminal anaesthesia at 15 min post administration of recombinant GM-CSF. Differential blood cell counts were analysed on an ADVIA120 (Bayer, Tarrytown, New York, USA).
The effects of GM-CSF-Rα inhibition in the collagen induced arthritis model of RA
Male DBA/1 mice (Jackson Laboratory, Bar Harbor, Maine, USA) were dosed with 100 μg bovine type II collagen in Freund's Complete Adjuvant at the base of the tail (two intradermal sites, 50 μL/site) followed by a subcutaneous injection of buprenorphine (0.1 mL/mouse) to induce arthritis. Onset of arthritis was determined as the 1st day that the clinical paw swelling score (range 0–4 per paw) was ≥1, typically 28±7 days post injection of collagen; each animal was then allocated alternately into the following treatment groups: vehicle; isotype control (CAT-004); CAM-3003 either 1 mg/kg of 10 mg/kg; or prednisolone (n=14/15 per group). Animals were dosed daily for 14 days and the clinical score was recorded daily.23
Mice were terminated at study end point and histological analyses of the rear ankle joints were performed to detect murine macrophages (clone Cl:A3-1, AbD Serotec, Kidlington, UK) (see online supplementary material for methods). All immunohistochemistry staining steps were carried out using a Dako AutoStainer (Glostrup, Denmark). Digital images were generated with an Aperio Scanscope XT scanning system (Vista, California, USA). The numbers of clearly distinguishable macrophages present within the annotated synovial membrane of the tibia-calcaneum were manually counted, up to an upper limit of 200 cells/mm2. The number of macrophages present was then recorded as total macrophage count.
All mice were housed in specific pathogen-free conditions at MedImmune, Cambridge, in compliance with Home Office Regulations (UK). All animal experiments were approved by the Animal Welfare and Ethics Committee (Babraham Institute).
Data analysis
Patient data: Continuous data were tested for normality and described as mean±SD or median and IQR as appropriate. A Mann–Whitney U test was performed to compare differences in GM-CSFRα positive cell counts between RA and PsA versus non-inflammatory controls (OA and HC).
Mouse model data: one way analysis of variance with Dunn's multiple comparison test was used to analyse joint score and histopathology counts from the collagen induced arthritis (CIA) model. The same statistical analyses were also used for the blood cell margination pharmacology model.
In all analyses a p value of <0.05 was considered statistically significant. All statistical analyses for in vitro and in vivo studies were performed using GraphPad PRISM 5 (GraphPad Software, La Jolla, California, USA).
Results
GM-CSFRα overexpression in synovial tissue of patients with RA and PsA
Clinical characteristics of study patients are described in table 1. The number of GM-CSFRα positive cells was significantly higher in ST from patients with active RA and PsA when compared with OA and HC (non-inflammatory controls) (figure1 A). There was no statistically significant difference in the number of GM-CSFRα positive cells in RA compared with PsA (p=0.79; data not shown). Immunofluorescent labelling demonstrated colocalisation with the macrophage markers CD68 and CD163 (figure 1B). No significant correlation was observed with disease activity parameters (disease activity score in 28 joints or C reactive protein) (data not shown).
Patient characteristics
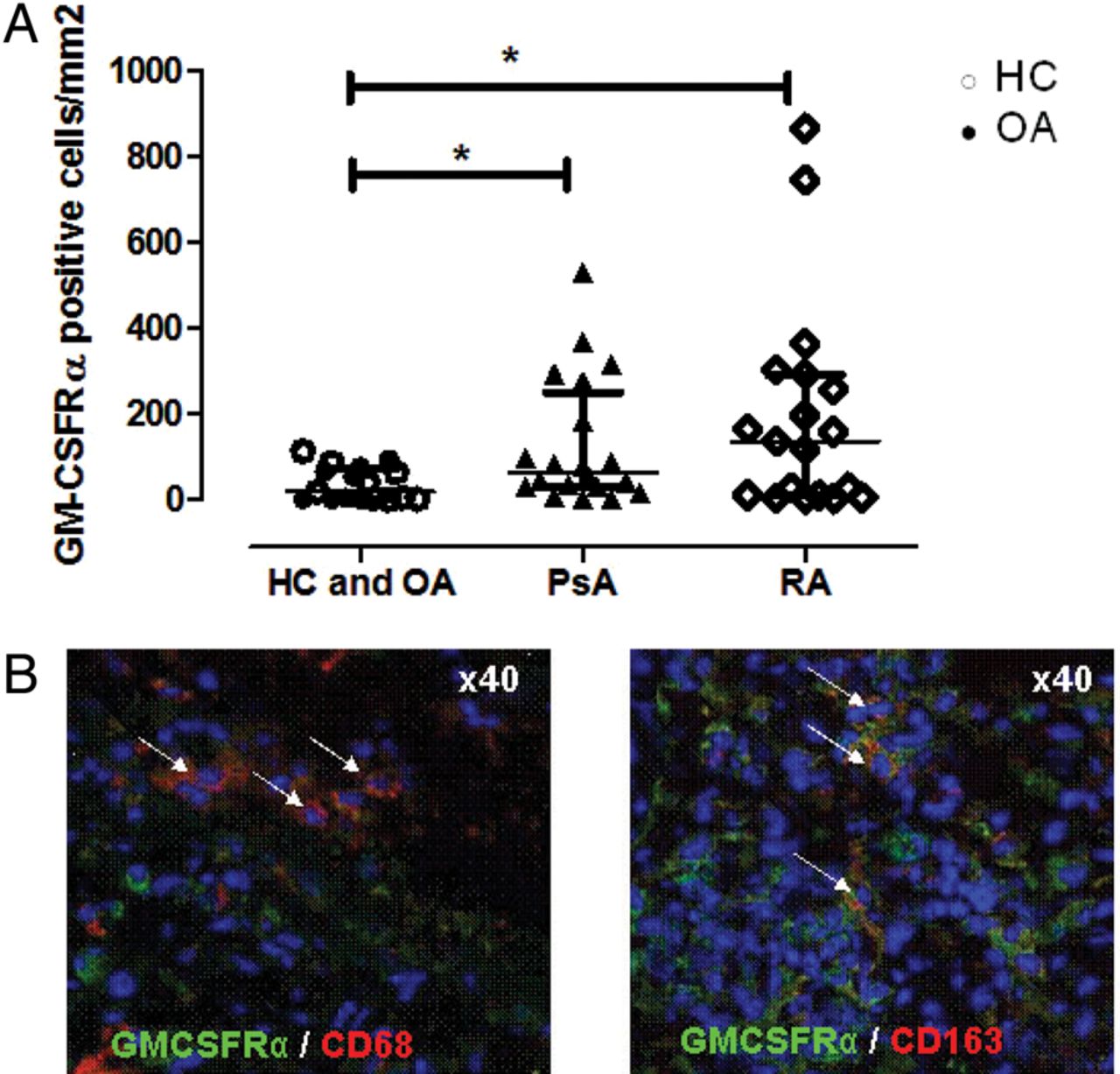
(A) The quantification of granulocyte macrophage colony stimulating factor (GM-CSFRα) positive cells in ST from patients with rheumatoid arthritis (RA) and psoriatic arthritis (PsA) (inflammatory arthritis; n=50) compared with ST from patients with osteoarthritis (OA) and healthy controls (HCs) (non-inflammatory; n=19). Median (IQRs) are shown. *p Value <0.05 (Mann–Whitney U test). (B) Representative picture of an immunofluorescence staining for GM-CSFRα (green). GM-CSFRα shows colocalisation with either CD68 (red) or CD163 (red) (arrows).
Pharmacokinetics of an antimouse GM-CSFRα chain antibody
To improve our understanding of GM-CSFRα inhibition a mouse GM-CSFR chain surrogate antibody (CAM-3003) was developed for in vivo pharmacology, as the clinical antibody mavrilimumab does not cross-react with rodents. From the Phase I study it was shown that mavrilimumab demonstrated non-linear pharmacokinetics14; therefore we explored the pharmacokinetics of the surrogate antibody. Analysis demonstrated that CAM-3003 had a non-linear pharmacokinetic profile (see online supplementary figure S1), similar to that observed with mavrilimumab, supporting its use as a relevant molecule for in vivo pharmacology.
Pharmacodynamic activity of GM-CSFRα inhibition in BALB/c mice
GM-CSF concentration-dependently increased CD11b expression on granulocytes (figure 2A) with a median effective concentration (EC50) of 430 pM (95% CI 385 pM to 499 pM). CAM-3003 dose-dependently inhibited GM-CSF induced CD11b upregulation (figure 2B) with a median inhibit concentration (IC50) of 150 pM (95% CI 124 pM to 204 pM). Next, we explored whether CAM-3003 could inhibit the margination of peripheral blood white blood cells to endothelial cells on the vessel walls following the administration of recombinant mouse GM-CSF. The number of circulating neutrophils (figure 3A) and monocytes (figure 3B), but not lymphocytes (figure 3C), decreased after 15 min and this was unchanged in the presence of isotype control (CAT-004). A complete inhibition was observed with administration of 10 mg/kg and 1 mg/kg CAM-3003, but not with 0.1 mg/kg. PBS alone or PBS and CAT-004 did not prevent this reduction in neutrophils (figure 3A) and monocytes (figure 3B). These data demonstrate that CAM-3003 can dose-dependently inhibit GM-CSF mediated margination of peripheral blood monocytes and neutrophils.

Characterisation of the cell surface expression of CD11b on mouse granulocytes stimulated with mouse granulocyte macrophage colony stimulating factor (GM-CSF). Mouse granulocytes were stimulated with a dose response of mouse GM-CSF for 1 hr and cell surface expression of CD11b quantified by flow cytometry using the mean fluorescent intensity (MFI). GM-CSF was able to dose dependently increase CD11b expression. (B) Mouse granulocytes were stimulated with mouse GM-CSF corresponding to the EC80 (2.5 ng/mL) and incubated with increasing concentrations of CAM-3003 for 1.5 h. Cells were then labelled for CD11b expression and the MFI quantified by flow cytometry. CAM-3003 dose-dependently inhibited GM-CSF induced CD11b upregulation.

(A) Characterisation of the in vivo neutralising activity of CAM-3003 in a model of granulocyte macrophage colony stimulating factor (GM-CSF) induced leucocyte margination. BALB/c mice were dosed intravenously with either CAM-3003 at 10 m/kg, 1 m/kg and 0.1 m/kg, the isotype control (CAT-004) at 10 mg/kg or vehicle (PBS) and then stimulated with mouse GM-CSF (mGMCSF, 0.25 μg) for 15 min. Peripheral blood was analysed using an ADVIA blood cell counter. (A) Absolute number of circulating neutrophils. (B) Absolute number of circulating monocytes. (C) Percentage of circulating lymphocytes 15 min post administration of GM-CSF. Statistically significant changes were observed due to changes in percentage of neutrophils and monocytes due to margination. Absolute numbers of lymphocytes did not change (data not shown). Data are expressed as mean±SEM (n=7–8). *p<0.05; ***p<0.001.
GM-CSFRα inhibition reduces disease severity in the CIA model of RA
To investigate the effect of GM-CSFRα inhibition, mice that were subjected to CIA were treated with CAM-3003 for 14 days after the first signs of arthritis. A statistically significant inhibition of clinical score was observed with maximal effect at 10 mg/kg intraperitoneally when compared with vehicle (figure 4A, B). This reduction in clinical score was equivalent to anti-GM-CSF and anti-tumour necrosis factor (TNF) inhibition in this model (see online supplementary figure S3). In addition, mice treated with CAM-3003 had less evidence of cellular infiltration, cartilage damage and bone erosion (figure 4C) and a significant reduction in the number of F4/80 positive macrophages when compared with isotype control (figure 4D). Splenomegaly was observed in arthritic animals and this was significantly reduced in mice treated with prednisolone and mice treated with 10 mg/kg of CAM-3003 (see online supplementary figure S4A). Thoracic and inguinal lymph nodes were pooled and weighed. Anti-GM-CSFR treated animals showed no reduction in lymph node weights in contrast to prednisolone treated animals (see online supplementary figure 4B). Peripheral blood cell counts did not show any significant difference in any white blood cell population in CAM-3003 treated mice (see online supplementary table S2) when compared with vehicle using a one way analysis of variance. However, in arthritic mice the absolute neutrophil count was significantly lower in mice treated with CAM-3003 10 mg/kg when compared with isotype treated mice (see online supplementary figure S5) using a t test. There was no statistically significant reduction in anti-type II collagen antibody levels between treatment groups (see online supplementary Figure S6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Characterisation of the effect of GM-CSFRα inhibition in established arthritis. (A) Mice were treated daily for 14 days post the onset of arthritis with either CAM-3003 at 10 mg/kg or 1 mg/kg, CAT-004 (10 mg/kg) as an isotype control, prednisolone (dose) or vehicle alone. Median clinical score was plotted daily to map disease progression. (B) Comparison of mean clinical score at Day 14 from three independent studies. Mean time of onset of arthritis was 28±7 days. Data is expressed as mean±SEM. (C) Histological images of mouse ankle joint from collagen induced arthritis model. Panel A–C are representative H&E images of (A) CIA induced arthritic joint treated with an isotype control antibody (CAT-004; 10mg/kg), (B) CIA induced arthritic joint treated with anti-GM-CSFRα antibody (CAM-3003; 10 mg/kg) (C) naive mouse joint (D) immunohistochemistry staining of a CIA induced arthritic mouse joint, demonstrating an infiltration of F4/80+ cells into the synovium. (D) Quantification of total numbers of F4/80+ cells located within the annotated area (see online supplementary figure S2) within the synovial membrane area. Data are from a collagen induced arthritis model treated with either an antimurine GM-CSFRα antibody (CAM-3003) or an isotype control antibody (CAT-004). Macrophages were assessed by positive membrane staining with anti-F4/80 immunohistochemistry. Data shown represent total F4/80+ counts for both hind paws of animals (n=14/15 per group) and are represented as mean±SEM. *p<0.05.
Discussion
The results presented here show an increased number of GM-CSFRα expressing cells in ST of patients with either RA or PsA when compared with non-inflammatory controls. GM-CSFRα expression in ST in RA and OA has been described before,12 ,24 but this is the first study to ascertain that GM-CSFRα positive cell numbers are higher in inflammatory arthritis versus non-inflammatory arthritis. In addition GM-CSFRα expression in PsA ST has not been described before. In addition, we have shown expression of GM-CSFRα by CD68 positive and CD163 positive macrophages. Of importance, the number of CD68 positive macrophages, one of the target cells for GM-CSF, correlates with disease activity in RA25 and the number of these cells reduce in the inflamed synovium following successful treatment.26 ,27 ,28 Together with the notion that the infiltration of the synovium by macrophages is similar between patients with RA and PsA,29 our results suggest that targeting GM-CSFRα could be effective in patients with RA and PsA.
Cell trafficking and retention of inflammatory cells play a key role in synovial inflammation. We found that mouse granulocytes incubated with CAM-3003, a GM-CSFR antagonist, could concentration-dependently inhibit GM-CSF induced upregulation of the integrin CD11b, which could indicate that blockade of GM-CSFRα can reduce synovial inflammation via blocking the actions of its ligand. Moreover, we showed with our mouse model that GM-CSFR blockade with CAM-3003 prevented the margination response of recombinant GM-CSF of white blood cells, consistent with observations in man30 ,31 and cynomolgus monkey.32 ,33 As we (data not shown) and others have shown that GM-CSF is elevated within the SF of patients with RA10 ,11 it appears likely that peripheral white blood cells coated in antibodies to the receptor would not respond to locally high concentrations of GM-CSF and therefore reduced margination and ultimately migration, diapedesis34 and retention within the joint would be expected to occur.
In line with the notion that anti-GM-CSFRα antibody treatment could have a beneficial effect on arthritis, we found in the murine CIA model of RA that CAM-3003 treatment dose-dependently inhibited clinical signs and symptoms of arthritis, with reduced synovial inflammation and joint destruction. This effect is probably due to inhibition of trafficking and retention of inflammatory cells, and secondary to other effects such as reduced survival of fully differentiated macrophages within the joint.1 These observations are supported by previous work demonstrating a reduction in inflammatory cell numbers in the joint35 and a reduction in Ly-6Chigh and Ly-6Clow monocytes in joint homogenates after GM-CSF blockade.36 A 14.7% reduction in spleen weight was also observed consistent with reduced inflammation in these animals. Splenomegaly has previously been described in a subset of patients with RA.37 The significance of this reduction has yet to be determined. There were no statistically significant changes in peripheral cell counts or body weight in mice with arthritis treated with the GM-CSFRα antibody throughout the study, however, whereas neutropenia was not observed, a slight reduction in neutrophils was observed in arthritic mice after CAM-3003 administration, suggesting that close monitoring of these cells in clinical studies with a GM-CSFRα inhibitor is warranted.
Taken together, these results further validate the GM-CSF–GM-CSFRα pathway as a new therapeutic target for the treatment of RA and perhaps PsA. Recent phase 1 and phase 2 clinical trials targeting either GM-CSF or GM-CSFRα13 ,14 ,15 support the development of new treatments interfering with this pathway.
Acknowledgments
The authors thank the following persons for their contribution to this manuscript: Academic Medical Center/University of Amsterdam: Alian Bakker, Maarten de Boer and Eva Ebbing for performing the synovial tissue stainings and Man-Wai Tang for data management.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
Footnotes
Handling editor Tore K Kvien
DEAG, ESC, MAS and PPT contributed equally.
Contributors MAS and PPT designed the study. Acquisition of the data was performed by DEAG, ESC, DMG, JC, JW, ND, AvN, AL, SH, MMC, DC, AD, EJ and GS. Data were analysed and interpreted by DEAG, DMG, IPW, IKA, AN, GS, MAS, PPT. The manuscript was drafted by DEAG and MAS and was revised by DMG, PPT. All authors read and approved the final manuscript. All authors agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding This work was partly funded by MedImmune; a wholly owned subsidiary of Astrazeneca. This work was also partly funded by CSL Ltd, Australia.
Competing interests PPT and DMG became employees of GlaxoSmithKline after study completion. GSK has not been involved in this study, but does have a proprietary interest in MOR103, an anti-GM-CSF antibody, by virtue of an alliance with Morphosys. ESC, ND, AL, SH, DC, AD, EJ and MAS are employees of MedImmune, a wholly owned subsidiary of Astrazeneca. IKA, AN, JC, JW and MMC were at the time of completing this work full-time employees of MedImmune, a wholly owned subsidiary of Astrazeneca. IPW received grants from CSL during the conduct of the study; grants from CSL, from null, outside the submitted work. MedImmune and Astrazeneca has an anti human GM-CSFRα chain antibody, mavrilimumab, in clinical development in rheumatoid arthritis.
Ethics approval Ethics committee of the AMC.
Provenance and peer review Not commissioned; externally peer reviewed.