Article Text

Abstract
Objective Human bone marrow mesenchymal stromal cells (hBM-MSC) are being applied in tissue regeneration and treatment of autoimmune diseases (AD). Their cellular and immunophenotype depend on isolation and culture conditions which may influence their therapeutic application and reflect their in vivo biological functions. We have further characterised the phenotype induced by fibroblast growth factor 2 (FGF2) on healthy donor hBM-MSC focusing on the osteoimmunological markers osteoprotegerin (OPG), receptor activator of nuclear factor kB (RANK), RANK ligand (RANKL) and HLA-DR and their regulation of expression by the inflammatory cytokines IL1β and IFNγ.
Methods RANK, RANKL, OPG and HLA-DR expression in hBM-MSC expanded under specific culture conditions, were measured by RT-PCR and flow cytometry. MAPKs induction by FGF2, IL1β and IFNγ in hBM-MSC was analysed by immunoblotting and RT-PCR.
Results In hBM-MSC, OPG expression is constitutive and FGF2 independent. RANKL expression depends on FGF2 and ERK1/2 activation. IL1β and IFNγ activate ERK1/2 but fail to induce RANKL. Only IL1β induces P38MAPK. The previously described HLA-DR induced by FGF2 through ERK1/2 on hBM-MSC, is suppressed by IL1β through inhibition of CIITA transcription. HLA-DR induced by IFNγ is not affected by IL1β in hBM-MSC, but is suppressed in articular chondrocytes and lung fibroblasts.
Conclusions RANKL expression and IL1β regulated MHC-class II, both induced via activation of the ERK1/2 signalling pathway, are specific for progenitor hBM-MSC expanded in the presence of FGF2. HLA-DR regulated by IL1β and ERK1/2 is observed on hBM-MSC during early expansion without FGF2 suggesting previous in vivo acquisition. Stromal progenitor cells with this phenotype could have an osteoimmunological role during bone regeneration.
- Cytokines
- Fibroblasts
- Inflammation
Statistics from Altmetric.com
Introduction
Human mesenchymal progenitor stromal cells (hMSC) isolated from bone marrow (BM) or other sources, have been studied in the laboratory for their multilineage cell differentiation potential and their ability to suppress lymphocyte proliferation.1 ,2 Due to these properties, mesenchymal stromal cells (MSC) are being investigated for therapeutic and commercial applications.3–8
In most cases, the hMSC are expanded in culture. Despite multiple cell divisions, they retain in vitro cell differentiation and antiproliferative properties. Fibroblast growth factor 2 (FGF2) is a pleiotropic growth factor involved in several cellular pathways during embryogenesis and in adult life.9 ,10 FGF2, although described as an inhibitor of cellular senescence,11 is also a potent cell mitogen. The addition of FGF2 to hBM-MSC cultures achieves high cell yields and favours enrichment of more progenitor cells.12
FGF2 participates in several bone metabolic processes, including skeletogenesis,13 osteocyte differentiation14 ,15 and osteogenic differentiation of MSC.16 FGF2 also selects MSCs best suited for ectopic bone formation.17
Healthy bone is a balance between bone formation by osteoblasts and bone resorption by osteoclasts. The alteration of this balance is a feature of some autoimmune diseases (AD) such as rheumatoid arthritis (RA) and of degenerative bone conditions. In the cells involved in bone metabolism, the expression of the receptor activator of nuclear factor kB (RANK), its ligand (RANKL) and osteoprotegerin (OPG), the RANKL decoy receptor, is central in bone remodelling by the immune system in health and disease.18 ,19 In the BM niche, stromal cells expressing OPG and RANKL participate in bone homeostasis as precursors of osteoblasts while supporting haematopoietic cell differentiation and osteoclasts precursors.20 In the BM, activated T cells, whether cytokine or antigen driven, express RANKL, whereas BM macrophages and osteoclasts express RANK.20–22 RANKL binding to RANK on preosteoclasts activates osteoclast maturation and osteoclastogenesis. Progenitor hBM-MSC constitutively express OPG.21 ,23
Inflammatory cytokines such as IL1β and interferon gamma (IFNγ) regulate bone homeostasis as mediators of cross talk between the bone and the immune system.18 In differentiated cells, IL1β can suppress IFNγ induced HLA-DR,24 and IFNγ can inhibit IL1β binding and signalling with implications for immunological responses and cytokine-induced tissue destruction.25
We26 ,27 and others28 ,29 have reported that in cultured hBM-MSC FGF2 induces expression of major histocompatibility complex (MHC)-classII (HLA-DR), a phenotypic marker of antigen presentation in haematopoietic and non-haematopoietic cells, constitutive in professional antigen-presenting cells and inducible by IFNγ in most cells. The induction of HLA-DR by FGF2 in vitro occurs only in hMSC (but not in mouse MSC) and requires the activation of ERK1/2 and cell proliferation. Neither of these conditions are required for the induction of HLA-DR by IFNγ.27 Expression of HLA-DR is observed also in some hBM-MSC during early in vitro expansion without FGF227 and could represent a residual in vivo FGF2 or IFNγ induced phenotype.
Although the HLA-DR on the hBM-MSC is functional, its significance when induced by FGF2 is not known. Adipose derived hMSC also respond to FGF2 with HLA-DR expression,27 but stromal cells from the Wharton's jelly of the umbilical cord do not express HLA-DR under induction of either FGF2 or IFNγ.30 ,31
In this study, we have investigated RANK, RANKL and OPG gene expression in hBM-MSC expanded in vitro with or without FGF2 and in the presence of IL1β and IFNγ. We also investigated whether BM-MSC HLA-DR expression induced by FGF2 and by IFNγ could be affected by IL1β.
Materials and methods
Cells, growth factors and cytokines
Human BM-MSC were obtained from healthy donors, and in vitro expanded as previously described in complete 10% fetal bovine serum (FBS) αMEM, defined as control (CTR) medium.27 To prevent senescence-related effects,32 the cells were not expanded for more than three passages. FGF2 (5 ng/mL, R&D) was added when stated.
IL1β (Sigma–Aldrich), IFNγ (R&D), IL1Ra (R&D) and the inhibitors UO126 (Sigma–Aldrich), PTDC (Sigma–Aldrich) and SB203580 (Cell Signaling Technology) were used as described in the figure legends.
RT-PCR
RT-PCR was performed as previously reported27 from cDNA isolated from hBM-MSC expanded under the conditions described in the figure legends. RT-PCR primers and probes for the human genes: β-actin, indoleamine 2,3-dioxygenase (IDO), CIITA, RANK, RANKL and OPG were purchased from Assays on Demand, Gene Expression products (Applied Biosystems).
Western blottting (WB)
BM-MSC were grown to semiconfluence in complete (10% FBS) αMEM, then maintained for 24 h in αMEM without FBS, before pulsing with FGF2, IL1β and IFNγ. Immunoblotting of whole cell lysates was performed as described previously.33 The following primary antibodies were used: rabbit antiphosphothr202/tyr204-ERK1/2, rabbit antiphosphoser473-Akt, rabbit antiphosphothr180/tyr182-p38MAPK (Cell Signaling Technology) and goat anti-GAPDH (Abcam, UK) as the internal protein loading control. Secondary HRP-conjugated goat antirabbit IgG (Southern Biotechnology) or donkey antigoat IgG (Santa Cruz Biotechnology) together with Amersham ECL (Amersham Biosciences, UK) were used to detect immunoreactive proteins. Scanned images of autoradiograms were analysed using AIDA Image or Scion (NIH) Image software.
Fluorescence-activated cell sorting (FACS)
This was performed as previously described.34 All antibodies were from BD (Switzerland).
Statistical analysis
Data are reported as the mean±SD. Comparisons between two groups were performed by Student 2-tailed t test.
Results
FGF2 induces RANKL expression via the ERK1/2 signalling pathway in hBM-MSC
The expression of OPG, RANKL and RANK was tested by RT-PCR in hBM-MSC (from five healthy donors) which had been expanded as follows: without FGF2 (CTR, figure 1A), in the continuous presence of FGF2 (FGF, figure 1B), or with addition of FGF2 after initial expansion under CTR conditions27 (CTR+F, figure 1C). OPG was constitutively expressed at similar levels under all three culture conditions. RANKL and RANK were expressed only in the presence of FGF2 although RANK was induced at much lower levels (≤10×) than RANKL (figure 1B,C). IL1β moderately increased OPG (figure 1A,B) and RANKL expression (figure 1B). In the absence of FGF2, IL1β was unable to induce either RANKL or RANK expression (figure 1A). FGF2 also induced CIITA in the CTR+FGF2 cells (figure 1C) as expected.27 In hBM-MSC expanded without FGF2, IFNγ did not induce RANK (figure 1A) nor significantly modified OPG and RANKL expression (not shown). UO126, an inhibitor of MEK1-235 (figure 2E), reduced significantly the expression of RANKL but not of RANK in FGF2 expanded hBM-MSC (figure 1D).
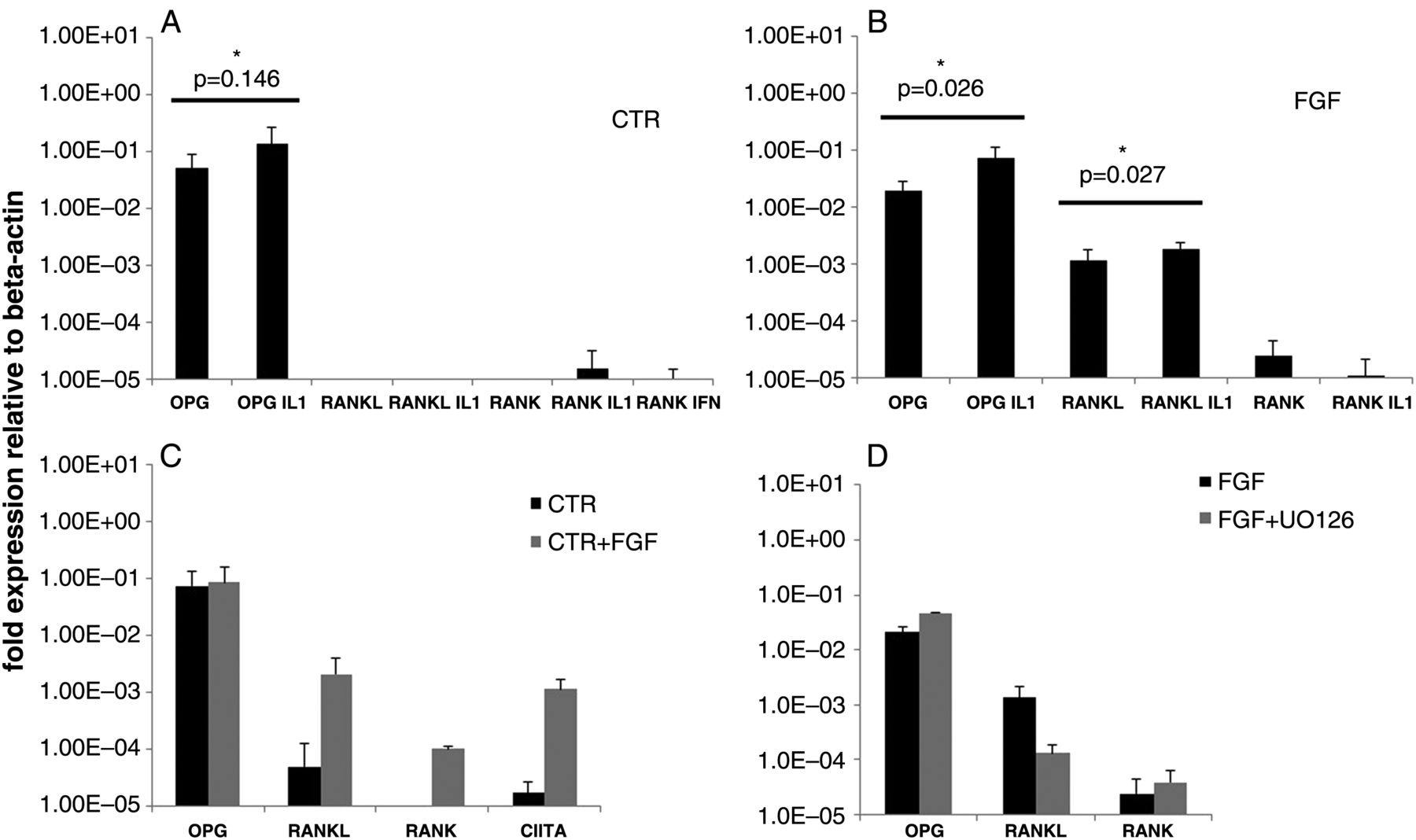
Modulation of receptor activator of nuclear factor kB (RANK), RANK ligand (RANKL) and osteoprotegerin (OPG) mRNA expression (measured by RT-PCR) in bone marrow mesenchymal stromal cells (BM-MSC) expanded: (A) without fibroblast growth factor 2 (FGF2) (CTR), (B) with FGF2(FGF)(5 ng/mL) or (C) without FGF2 as in (A) with successive addition of FGF2(CTR+FGF). Incubation with FGF2 (in CTR+F, (C)) and IL1β(IL1)(1 ng/mL) or IFNγ(IFN)(10 ng/mL) was for 96 h. (D) Effect of the MEK1-2 inhibitor UO126(10 μM) on RANK, RANKL and OPG mRNA expression in FGF2-expanded BM-MSC as in (B). Cells were incubated with UO126 for 96 h. Each histogram represents the relative expression of each gene under every cell culture condition. *p<0.05=significant.

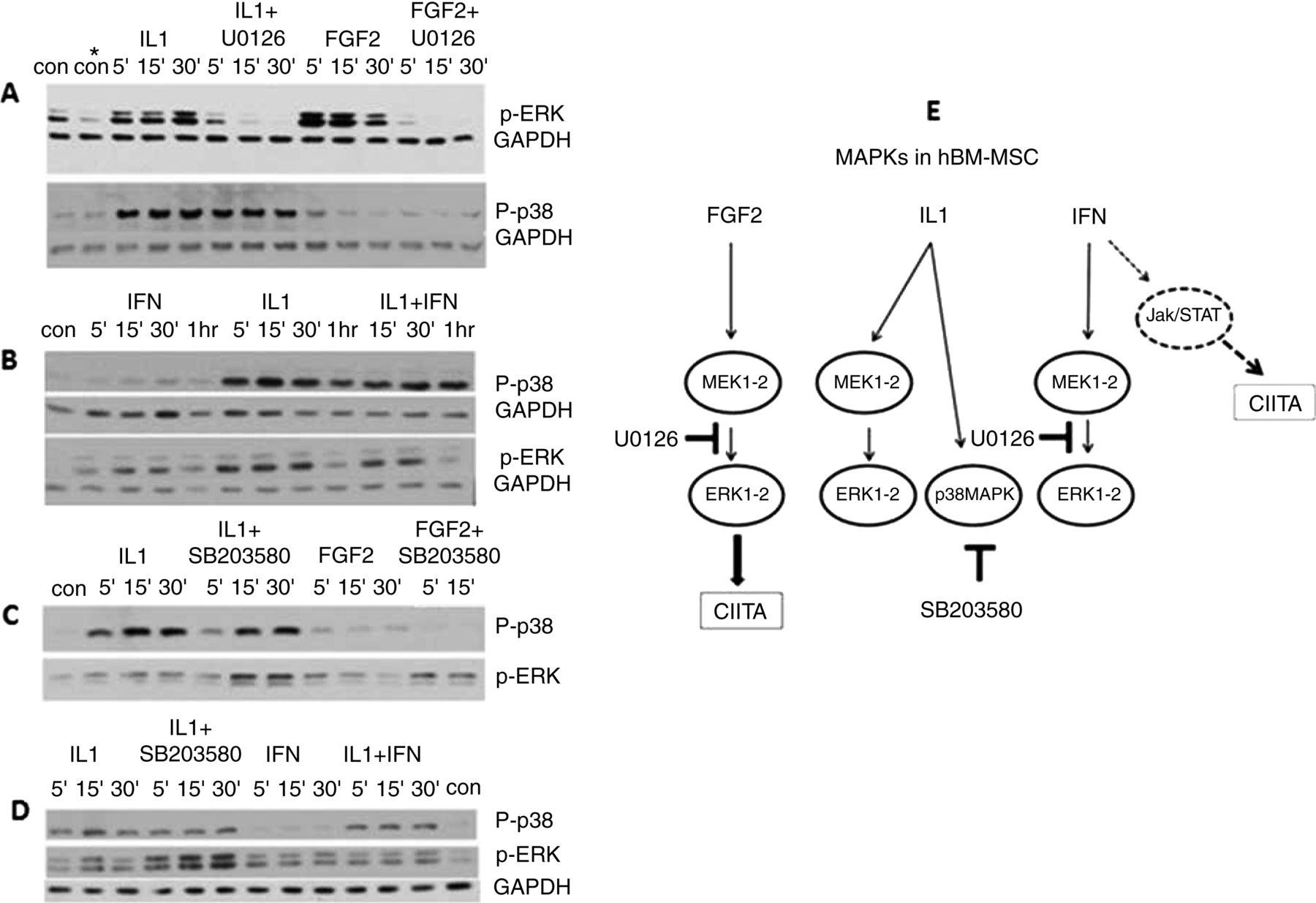
(A–D) WB analysis of the MAPK proteins (ERK1/2 and p38) induced in bone marrow mesenchymal stromal cells (BM-MSC) by fibroblast growth factor 2 (FGF2) (5 ng/mL), IL1β(IL1)(1 ng/mL) and IFNγ(IFN)(10 ng/mL). UO126: MEK1-2 inhibitor (10 μM), SB203580: p38 inhibitor (10 μM); P-p38: phosphorylated p38 (MW: 40 kDa), P-ERK1/2: phosphorylated ERK1-2 (MW: 42–44 kDa). GAPDH (MW: 35 kDa): internal protein control. Time intervals of exposure to each factor are indicated above each panel. In experiments with kinase inhibitors, the cells were preincubated with these (4–18 h) before being pulsed with either FGF2 or the cytokine. (E) Schematic diagram of the MAPKs activated by FGF2, IL1 and IFN in BM-MSC and of the inhibitory activity of UO126 or SB203580 in the ERK1/2 signalling pathway. Elements drawn in broken line represent published data not included in this study.
IL1β lowers CIITA transcription and HLA-DR expression induced by FGF2 in hBM-MSC
Addition of IL1β during hBM-MSC expansion (from five donors) with FGF2, reduces the expression of HLA-DR (figure 2A). The IL1β doses chosen, (50–1000 pg/mL), which are physiologically relevant, have previously been shown to induce in vivo and in vitro mineralisation enhancement during osteogenic differentiation.36 Competition for IL1R1 binding with IL1Ra, the IL1R1 antagonist, shows that IL1β reduces HLA-DR by binding to its own receptor (figure 2B). Intercellular adhesion molecule (ICAM) (CD54), important in immunological intercellular interactions, is expressed in hBM-MSC independently of FGF2 and is upregulated by IL1β (figures 2C and 3C). IL1Ra inhibits the upregulation of CD54 by IL1β, confirming the specific binding of IL1β to IL1R1. Expression of FGFR1, the main FGF2 receptor on these cells under these conditions,27 is not downregulated by IL1β (figure 2F). Cell proliferation, a necessary condition for the maintenance of FGF2-induced HLA-DR,27 is not reduced.

HLA-DR expression, measured by FACS, in fibroblast growth factor 2 (FGF2) expanded bone marrow mesenchymal stromal cells (BM-MSC) (FGF) in presence of (A) IL1β(IL1), (B) IL±IL1Ra, (D) IL1±MEK1-2 inhibitor UO126 or (E) IL1±NFkB inhibitor PTDC. (C) CD54 (ICAM) expression, measured by FACS, in FGF BM-MSC in presence of IL1±IL+Ra. Filled grey histograms are control IgG. RT-PCR analysis of relative expression of (F) FGF2 receptor 1(FGFR1) and (G) CIITA in FGF BM-MSC±IL1±UO126 or PTDC. Each histogram represents the relative expression of each gene under every cell culture condition. FGF2: 5 ng/mL; IL1: 1 ng/mL; IL1Ra: 1–10 ng/mL; UO126, PTDC: 10 μM. Incubation times in all cases was for 96 h. *p<0.05=significant.

Effect of IL1β(IL1) on HLA-DR expression, measured by FACS, in human bone marrow mesenchymal stromal cells (hBM-MSC) expanded without fibroblast growth factor 2 (FGF2) (CTR condition), to which (A) IFNγ(IFN)(5–10 ng/mL) or (B) FGF2(CTR+FGF)(5 ng/mL) were subsequently added with or without IL (1–5 ng/mL). Simultaneous incubation with each cytokine and FGF2 (in CTR+F) was for 96 h. Alternatively, the cells were preincubated with IL1 for 24 h before addition of IFN. (C) CD54(ICAM) expression in BM-MSC with or without IL1±IFN. Filled grey histograms are control IgG. (D) Relative expression, measured by RT-PCR, of indoleamine 2,3-dioxygenase (IDO) enzyme in: FGF2 expanded BM-MSC alone(FGF) or with IL1 addition (FGF IL), and BM-MSC expanded without FGF2 (CTR), to which IFN (CTR IFN) or IFN+IL1 (CTR IL1 IFN) were added for 96 h at concentrations as above.
CIITA expression is necessary for the induction of HLA-DR in all cells. We have previously shown that this is the case also when HLA-DR is induced by FGF2 in hBM-MSC.27 IL1β significantly inhibits CIITA expression in these cells (figure 2G).
The expression of CIITA by FGF2 in hBM-MSC occurs through the activation of the ERK1/2 signalling pathway.27 Inhibition of MEK1-2/ERK1/2 with UO126 significantly reduces the expression of CIITA (figure 2G) and HLA-DR (figure 2D). Simultaneous addition of IL1β and UO126 further reduced CIITA transcription and HLA-DR expression (figure 2G,D), while simultaneous addition of PTDC, an inhibitor of NF-Kb, induced by IL1β in most cells, did not further reduce either CIITA (figure 2G) or HLA-DR (figure 2E) expression.
IL1β lowers neither HLA-DR nor indoleamine 2,3-dioxygenase (IDO) expression induced by IFNγ in hBM-MSC
Induction of HLA-DR by IFNγ in hBM-MSC occurs via activation of the Jak/Stat signalling pathway,37 does not require cell proliferation27 and is independent of MEK1-2/ERK1/2.27 IFNγ induces in hBM-MSC also IDO, an inhibitor of lymphocyte proliferation and, possibly, a mediator of the immunosuppressive effect of the hBM-MSC.38
We queried whether IL1β would suppress the induction of HLA-DR and of IDO by IFNγ in hBM-MSC. The IFNγ concentrations used (5–10 ng/mL) were chosen for their physiological significance, consistent with already published literature.24 ,27 The addition of IL1β and IFNγ, either simultaneous or with addition of IFNγ after preincubation with IL1 β, to hBM-MSC (from five different donors) expanded without FGF2 in CTR conditions, did not lower the HLA-DR induced by IFNγ, as exemplified in figure 3A. IL1β alone did not induce any HLA-DR (not shown). When FGF2 rather than IFNγ was added to the same cells, HLA-DR was expressed and suppressed by IL1β as expected (figure 3B). RT-PCR experiments showed also no reduction of CIITA expression due to IFNγ in the presence of IL1β (data not shown). The failed suppression by IL1β of IFNγ-induced HLA-DR was not due to the lowering by IFNγ of the expression of the IL1β receptor IL1R1, since CD54, upregulated by either IL1 or IFNγ, was synergistically increased when both cytokines were present (figure 3C).
Additionally, IL1β does not suppress IFNγ-induced expression of the IDO enzyme (figure 3D) and, therefore, the subsequent BM-MSC associated lymphocyte antiproliferative effect.
MAPKs activated in BM-MSC by IL1β, FGF2 and IFNγ
IL1β is recognised to activate members of the MAPK family of serine/threonine kinases, and in a cell context manner.39 Activation of the ERK1/2 signalling pathway is required by IL1β for in vitro osteogenic differentiation of hBM- MSC and to upregulate mineralisation genes,36 and by FGF2 to induce CIITA/HLA-DR and RANKL. At the same time, IL1β suppresses CIITA expression induced by FGF2 as shown above. One publication using WB analysis, shows that IL1β does not activate ERK1/2 in hBM-MSC.40 To our knowledge, no data exist with respect to the MAPKs induced by IFNγ in hBM-MSC. Therefore, we investigated by WB analysis whether the ERK1/2, p38 and JNK MAPKs were activated by FGF2, IL1β and IFNγ in hBM-MSC. Figure 4 shows representative results from six experiments, which were consistent, although quantitative variations occurred in the amount of phosphorylated kinase induced each time due to donor and cell culture variations.
FGF2, IL1β and IFNγ activate ERK1/2
JNK activation was not induced by either FGF2 or IL1β (data not shown). Its induction by IFNγ was not tested. Figure 4 shows that FGF2, IL1β (4A) and IFNγ (4B,D) all activated ERK1/2 within 5 min of addition to the cells. The presence of the MEK1-2 inhibitor UO126 severely impaired ERK1/2 phosphorylation induced by both FGF2 and IL1β, as expected (figure 4A).
Only IL1β activates p38MAPK
Phosphorylation of the p38MAPK was induced by IL1β (figure 4A–D) but not FGF2 (figure 4A,C) and IFNγ (figure 4B,D). IFNγ does not affect p38-induced phosphorylation induced by IL1β. SB203580 inhibits only the catalytic activity of phosphorylated p38, but not its phosphorylation by upstream kinases.41 However, SB203580 increases the amount of phosphorylated pERK1/2 induced by IL1β or FGF2 (figure 4C,D).
The HLA-DR expressed on BM-MSC expanded without FGF2 is suppressed by IL1β and the MEK1-2 inhibitor UO126
HLA-DR positive (≤5–10%) hBM-MSC have been observed in about 10% of the donors, also at the earliest expansion passage without FGF2. Such positivity disappears with further passaging or increases if FGF2 is added.27 Whether such positivity derives from previous in vivo exposure to IFNγ or FGF2 is not known. However, the discriminatory effect of MEK1-2 inhibition and IL1β on the HLA-DR induced by either factor should provide now some clarity.
BM-MSC from three healthy donors (32–50 years old) showed at the first expansion passage in the absence of FGF2 (or IFNγ) a significant HLA-DR positivity (20–30% positive cells), which diminished over successive expansion passages.
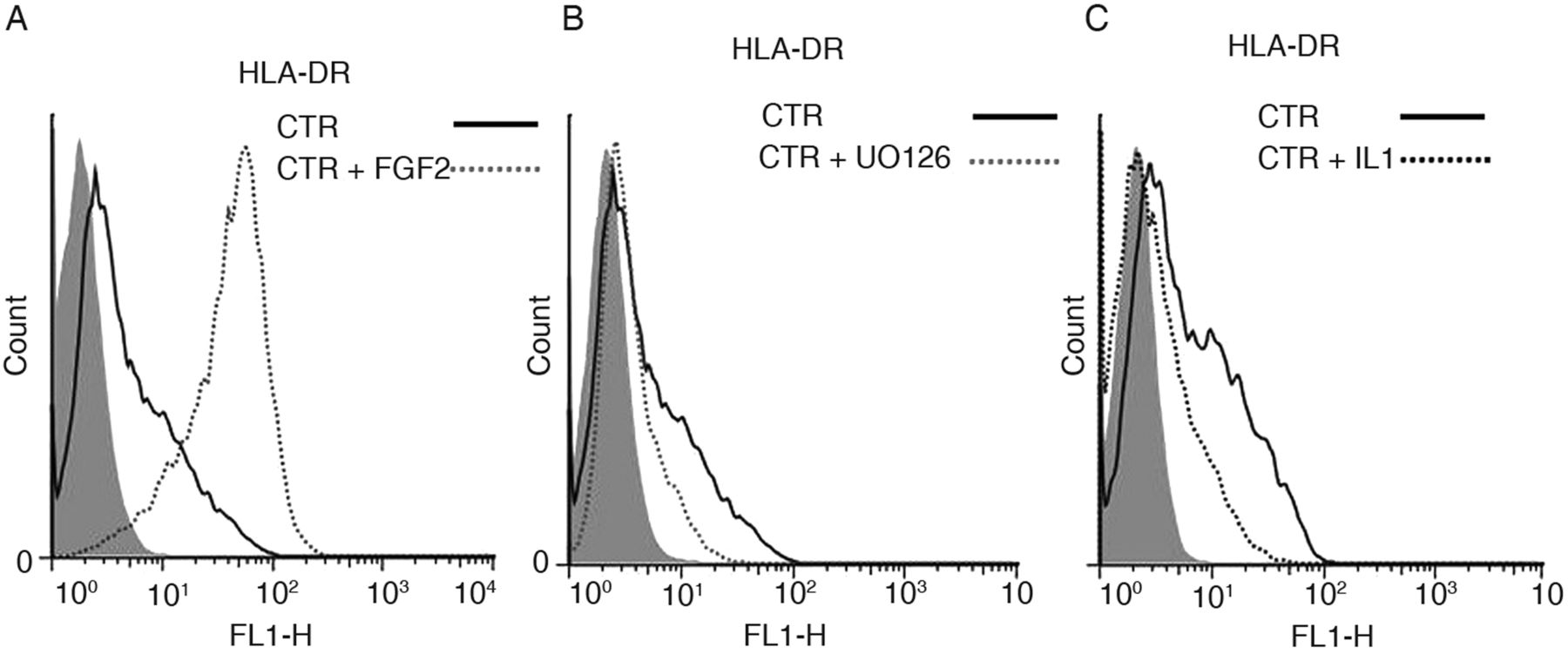
Addition of FGF2 amplified the original HLA-DR positive population (figure 5A) which was suppressed by both UO126 and IL1 β as exemplified in figure 5B,C respectively. These results suggest that the original HLA-DR was induced via the ERK1/2 signalling pathway and was not due to IFNγ.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
HLA-DR expression, measured by FACS, in human bone marrow mesenchymal stromal cells (hBM-MSC) expanded without fibroblast growth factor 2 (FGF2) (CTR condition), to which (A) FGF2, (B) the MEK1-2 inhibitor UO126 and (C) IL1β(IL1) were subsequently added. In (C) and (A): same donor CTR BM-MSC but at different expansion passage and different baseline HLA-DR expression. Filled grey histograms are control IgG.
Discussion
Although hBM-MSC have rapidly gone from the bench to the patient bedside for the treatment of a variety of AD and tissue degenerative conditions, knowledge as to their biology and the mechanisms underlying their functions is incomplete. The definition of the progenitor phenotype of the cells which are infused in vivo is arbitrary and dependent on the isolation method, culture conditions and cell source.
In view of the above translational applications, the osteoimmunological profile of the cells will be critical. This will depend on the combination of cytokines and growth factors encountered in the eventual inflammatory niche.
We26 ,27and others37 have reported that hBM-MSC expanded with FGF2 express HLA-DR. This HLA-DR is functional.27
The OPG/RANKL/RANK axis and its modulation is pivotal in bone formation in health and disease and is crucial for the development of new drugs against bone-related disorders.42
Human osteoblasts, but not BM-MSC, express RANKL.21 ,23 However, BM-MSC produce constitutively the decoy receptor of RANKL, OPG, which competes with RANK for RANKL binding thereby controlling RANKL-induced osteoclastogenesis.23 We show for the first time that hBM-MSC express RANKL and, to a much lower level, RANK, when FGF2 is added to the cells (figure 1B,C). RANKL, but not RANK, expression depends on ERK1/2 activation by FGF2 (figure 1D). Induction of RANKL/RANK expression was observed by others in hBM-MSC exposed to conditioned medium from activated T cells21 suggesting a role of inflammatory T cells in osteoblastic differentiation. Annexin II, a factor promoting osteoclast formation, has been reported also to stimulate in hBM-MSC RANKL mRNA and protein via ERK1/2.43 The significance, if any, of low RANK mRNA expression in hBM-MSC is unknown. We confirm that OPG expression is constitutive and unaffected by FGF2. FGF2-induced RANKL mRNA is moderately increased by IL1β (figure 1B), a cytokine often associated with osteoclastogenesis and bone erosion. IFNγ, which exerts an antiosteoclastogenic activity,44 does not significantly affect expression of either OPG or RANKL (data not shown).
In hBM-MSC, in parallel with the induction of RANKL, FGF2 induces via ERK1/2 also the expression of CIITA and HLA-DR.27 We then asked whether IL1β, secreted by monocytes, osteoclast precursors and in inflammation, would affect the expression of HLA-DR induced by either FGF2 or IFNγ.
We report for the first time that, in hBM-MSC, IL1β specifically inhibits CIITA transcription and HLA-DR expression only when FGF2-induced. IL1β suppresses neither HLA-DR (figure 3A) nor IDO enzyme expression induced by IFNγ (FGF2 induces no IDO expression in BM-MSC) (figure 3D), but upregulates CD54 (ICAM) in both FGF2 and IFNγ treated cells (figure 3C).
The ERK1/2 signalling pathway appears central for the induction of RANKL and CIITA by FGF2 in hBM-MSC. It mediates also the upregulation by IL1β of mineralisation genes in osteogenic differentiations.36 We therefore investigated by immunoblotting the MAPKs activated by FGF2, IL1β and IFNγ in hBM-MSC. We show for the first time that FGF2, IL1β and IFNγ all activate ERK1/2 (figure 4A–D). Yet, only FGF2 induces RANKL and CIITA expression via ERK1/2; IFNγ does not require ERK1/2 for CIITA/HLA-DR expression.27
Among FGF2, IL1β and IFNγ, only IL1β activates p38MAPK (figure 4C). In hBM-MSC, inhibition of cellular p38MAPK results in increased levels of pERK1/2 activated by FGF2 (figure 4C,D) or endogenous (not shown). Yet, in FGF2 expanded hBM-MSC, inhibition of cellular p38MAPK does not affect CIITA mRNA, but lowers HLA-DR expression. This occurs also when HLA-DR is induced by IFNγ in cells expanded without FGF2 (see online supplementary figure S2, supplementary data). These data suggest that p38 controls HLA-DR expression in hBM-MSC at an unidentified post-CIITA transcriptional level. P38 alone, when activated by IL1β, cannot therefore be responsible for the lowering of ERK1/2 activation dependent CIITA transcription. On the other hand, the latter is suppressed by IL1β in synergy with the inhibition of MEK1-2 (by UO126, figure 2G) and of p38 (by SB203580, online supplementary figure S2B, supplementary data), suggesting an additional control by IL1β on the CIITA transcription induced by FGF2 in hBM-MSC.
By inducing RANKL mRNA, FGF2 seems to skew the hBM-MSC toward an osteoblastic phenotype. The characterisation of the correspondent RANKL protein with respect to conditions of expression, cellular location and ratio to OPG, is necessary for better evaluating its biological and translational significance. These results, however, suggest caution in the use of hBM-MSC as therapeutic tools in the treatment of inflammatory disorders involving bone loss, such as RA.23 ,45 They could express RANKL in vivo in the presence of either FGF2, which is ubiquitously present especially during new bone and vascular formation, or of inflammatory lymphocytes.21 This effect of FGF2 should be investigated also on adipose tissue-derived MSC, which do require FGF2 for their expansion in vitro46 and are often used in translational applications.
In hBM-MSC, the expression via ERK1/2 of RANKL and CIITA/HLA-DR are specific to FGF2. Platelet derived growth factor (PDGF) type BB, often simultaneously present in vivo with FGF2, also induces HLA-DR in hBM-MSC,27 but has not been tested either for RANKL induction or HLA-DR sensitivity to IL1β.
Mitogenic proliferation via ERK1/2 activation, is necessary for HLA-DR expression induced by FGF2; whether it is necessary also for the expression of RANKL is not known. Although in the presence of FGF2, CIITA/HLA-DR and RANKL are simultaneously expressed and decline in parallel with cell passaging, there is no causal relationship between them because CIITA gene silencing with shRNA does not inhibit RANKL induction by FGF2 (unpublished results).
The expression of RANKL and IL1β-regulated CIITA/HLA-DR on progenitor hBM-MSC proliferating under the effect of FGF2, could represent the osteoimmunological phenotype of BM-MSC populations building cell mass at a specific phase of bone regeneration. T cells activated by antigen receptor engagement express RANKL and influence bone metabolism.47 The HLA-DR/RANKL/OPG expressing hBM-MSC could interact with the CD4 T-cells, and influence the osteoblasts/osteoclasts balance. HLA-DR on hBM-MSC due to either FGF2 or IFNγ is equally functional,27 and its discriminate suppression by IL1β may reflect different temporal and/or cell differentiation stages. In differentiated cells of mesodermal origin, chondrocytes and fibroblasts, FGF2 cannot induce CIITA/HLA-DR27 and IL1β suppresses HLA-DR due to IFNγ (see online supplementary figure S1A and B, supplementary data).
Finally, the hypothesis that HLA-DR positive hBM-MSC may occur in vivo, has been suggested by reports that non-expanded (though selected for a stem cell marker expression) hBM-MSC are HLA-DR positive but lose their positivity on passaging in culture.27 ,28 Our results showing that the HLA-DR positivity found on some hBM-MSC expanded without FGF2 requires the activation of the ERK1/2 (figure 5B) signalling pathway and is sensitive to IL1β (figure 5C), further support this hypothesis.
Acknowledgments
We thank Professor Ivan Martin, Department of Surgery and Biomedicine, University Hospital Basel, for his constructive discussions and logistics support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.