Article Text

Abstract
Objectives The safety and efficacy of sirukumab, an anti-interleukin-6 (IL-6) monoclonal antibody, were evaluated in a 2-part, placebo-controlled phase II study of patients with active rheumatoid arthritis (RA) despite methotrexate therapy.
Methods In Part A (proof-of-concept), 36 patients were randomised to placebo or sirukumab 100 mg every 2 weeks (q2w) through week 10, with crossover treatment during weeks 12–22. In Part B (dose finding), 151 patients were randomised to sirukumab (100 mg q2w, 100 mg q4w, 50 mg q4w, or 25 mg q4w) through week 24, or placebo through week 10 with crossover to sirukumab 100 mg q2w (weeks 12–24). The proportion of patients with an American College of Rheumatology 50 (ACR50) response and the change from baseline in the 28-joint count disease activity score using C-reactive protein (DAS28-CRP) were determined. Safety was evaluated through week 38 in both parts.
Results The primary endpoint (ACR50 at week 12 in Part B) was achieved only with sirukumab 100 mg q2w versus placebo (26.7% vs 3.3%; p=0.026). Greater improvements in mean DAS28-CRP at week 12 were observed with sirukumab 100 mg q2w versus placebo in Parts A (2.1 vs 0.6, p<0.001) and B (2.2 vs 1.1; p<0.001). The incidence of adverse events (AEs) was similar for sirukumab-treated and placebo-treated patients through week 12 in Part A (70.6% and 63.2%, respectively) and B (67.8% and 66.7%, respectively). Infections were the most common type of AE; one death occurred (Part B, sirukumab 100 mg q2w, brain aneurysm).
Conclusions Sirukumab-treated patients experienced improvements in the signs/symptoms of RA. Safety results through 38 weeks were consistent with other IL-6 inhibitors.
Trial registration number NCT00718718.
- Rheumatoid Arthritis
- Methotrexate
- Treatment
- DMARDs (biologic)
- Cytokines
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Interleukin (IL)-6 is a key mediator in the inflammatory process of rheumatoid arthritis (RA)1 and has been found at elevated levels in the serum, synovial tissue, and synovial fluid of patients with RA.2–5 Thus, IL-6 is an attractive target for new RA therapies, including patients who have had an inadequate response to or intolerance of antitumour necrosis factor (TNF) agents. Currently, tocilizumab, a humanised antibody targeting the IL-6 receptor, is the only approved therapy for RA that inhibits the IL-6 pathway.6 The efficacy and safety of binding the IL-6 ligand, rather than the IL-6 receptor, is not yet sufficiently clear.
Sirukumab (formerly known as CNTO 136) is a human anti-IL-6 monoclonal antibody that binds IL-6 with high affinity and specificity, thereby inhibiting IL-6-mediated effects.7 We report here the results of a 2-part, phase II study evaluating the safety and efficacy of sirukumab in patients with active RA despite methotrexate (MTX) therapy.
Methods
Patients
Adult patients (aged ≥18 years; ≥20 years at Japanese sites) with a diagnosis of RA8 for ≥4 months, active disease (≥6 swollen/≥6 tender joints), a serum C-reactive protein (CRP) level ≥10.0 mg/L, and a positive anti-cyclic citrullinated peptide antibody or rheumatoid factor status were enrolled. All patients were to have received MTX therapy (≥15 mg/week; ≥8 mg/week at Japanese sites only) for ≥4 months, with a stable dose for ≥6 weeks. Treatment with stable doses of sulfasalazine, hydroxychloroquine, or chloroquine in addition to MTX was allowed. Patients treated with stable doses of oral glucocorticoids (≤10 mg/day prednisone or equivalent) or nonsteroidal anti-inflammatory drugs (NSAIDs) were eligible, and continued on the same dose through week 24. Previous use of TNF inhibitors, tocilizumab, disease-modifying anti-rheumatic drugs (DMARDs) other than those noted above, or cytotoxic drugs was prohibited. Patients were also excluded from the trial if they had any signs or symptoms of severe, progressive, or uncontrolled renal, hepatic, haematologic, gastrointestinal, endocrine, pulmonary, cardiac, neurologic, or cerebral disease.
The protocol (NCT00718718) was approved by the local institutional review boards or ethics committees. All patients provided written, informed consent before study-related procedures were performed.
Study design
This was a 2-part, phase II, multicenter (Part A: 8 sites; Part B: 36 sites; Europe, North America, and Asia), randomised, double-blind, placebo-controlled study evaluating the efficacy and safety of sirukumab in patients with active RA despite MTX therapy. Different cohorts of patients were enrolled into Parts A and B. In both parts, randomisation was performed using an interactive voice response system. In order to achieve the desired assignment proportions within each stratum, defined by investigational site and weight group, an adaptive randomisation procedure with the minimisation algorithm based on biased-coin assignment9 was used in both parts.
In the proof-of-concept Part A, patients stratified by investigational site and weight group (< or ≥75 kg) were randomised (1:1) to subcutaneous (SC) placebo or sirukumab 100 mg every 2 weeks (q2w) through week 10, followed by crossover (placebo→sirukumab or sirukumab→placebo) during weeks 12–22. An interim analysis of the change from baseline in 28-joint count disease activity score using CRP (DAS28-CRP) and safety findings was conducted at week 12, and these results supported the initiation of Part B. In the dose-finding Part B, a separate cohort of patients stratified by investigational site and weight group (<65, 65−85, >85 kg) were randomly assigned (1:1:1:1:1) to receive SC sirukumab 100 mg q2w, 100 mg q4w, 50 mg q4w, or 25 mg q4w through week 24, or SC placebo q2w with crossover at week 12 to sirukumab 100 mg q2w through week 24. In both parts, patients were to continue their stable baseline dose of MTX through week 24, except when dosage adjustments were required due to MTX toxicity. Patients also received a stable dose of oral folic/folinic acid (≥5 mg/week) to reduce MTX-related toxicity.
Study assessments
In Parts A and B, the last study agent administrations occurred at weeks 22 and 24, respectively, with the final efficacy evaluations occurring at weeks 24 and 38, respectively. The primary endpoint of the study was the proportion of patients with ≥50% improvement in the American College of Rheumatology criteria (ACR50 response) at week 12 in Part B. Major secondary endpoints included ACR50 response at week 12 in Part A and changes from baseline in DAS28-CRP at week 12 in Parts A and B. Additionally, the proportions of patients with an ACR20 response,10 a good or moderate DAS28-CRP EULAR response,11 and DAS28-CRP remission (score <2.6) were also determined. Physical function and health-related quality of life were assessed with the health assessment questionnaire-disability index (HAQ-DI)12 and the physical/mental component summary (PCS/MCS) scores of the 36-item short form health survey (SF-36),13 respectively. Posthoc analyses determined the proportions of patients achieving remission based on clinical disease activity index (CDAI) ≤2.8,14 simplified disease activity index (SDAI) ≤3.3,15 and Boolean-based ACR/EULAR criteria15 as well as change from baseline in CDAI at weeks 12 and 24.
Safety evaluations were performed and adverse events (AE) were monitored through week 38 in Parts A and B. Serial serum samples were collected to evaluate serum sirukumab concentrations and the presence of antibodies to sirukumab. Pharmacokinetic parameters for sirukumab were calculated using a non-compartmental analysis method implemented in WinNonlin (V.5.2.1; Pharsight Corporation, Mountain View, California, USA). Additional samples were collected to evaluate levels of CRP and fasting lipids.
Statistical analyses
All analyses were performed separately for Parts A and B. Efficacy data were analysed by randomised treatment group. All patients who received ≥1 sirukumab administration were included in the safety analysis; safety data were reported using descriptive statistics by the treatment actually received. For efficacy analyses, patients who initiated prohibited medications, increased the dose of permitted medications, or discontinued study treatment due to unsatisfactory therapeutic effect prior to week 12 in both parts were considered treatment failures. Additionally, the last observation carried forward methodology was used to impute missing postbaseline data. Patients from one site in Part A (n=5; placebo, 2; sirukumab, 3) were excluded from all efficacy, pharmacokinetic and pharmacodynamic analyses due to concerns about data integrity; however, these patients were included in the safety analysis.
Differences between each sirukumab group and the respective placebo group in Parts A and B were assessed using a Cochran–Mantel Haenszel χ2 test stratified by weight (Part A: <75, ≥75 kg; Part B: <65, 65–85, >85 kg) for discrete variables and an analysis of variance on the van der Waerden normal scores stratified by weight (Part A: <75, ≥75 kg; Part B: <65, 65–85, >85 kg) for continuous variables. No multiplicity adjustment was applied for Part A. A sequential analysis was used in Part B, in which each sirukumab dose group was compared with the placebo group in the following order: 100 mg q2w, 100 mg q4w, 50 mg q4w, and 25 mg q4w. If a given treatment group comparison was not statistically significant, the remaining treatment group comparisons were not tested and were defined as being not significant.
A planned sample size of 20 patients/group for Part A provided ≥80% power to detect a difference of 1.0 in change from baseline in DAS28-CRP, assuming a SD of 1.2 using a one-sided t test at a significance level of 0.05. In Part B, a planned sample size of 30 patients/group provided ≥80% power to detect a treatment difference of 35% between placebo and sirukumab groups in week 12 ACR50 response rates using a two-sided χ2 test at a significance level of 0.05.
Results
Patient characteristics
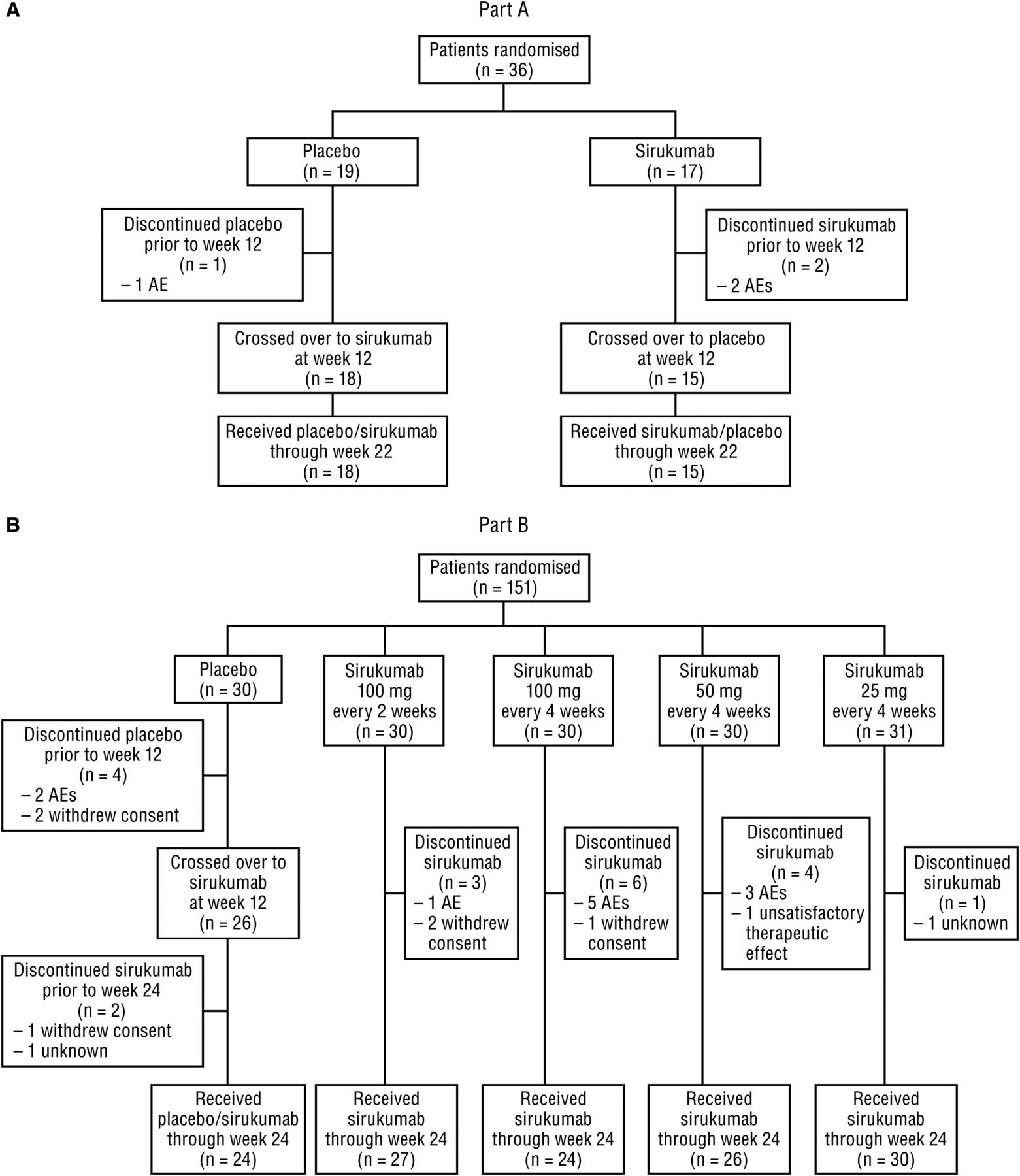
Data were collected from July 2008 through March 2011. Patient disposition is shown in figure 1. In Part A, 36 patients (placebo, n=19; sirukumab, n=17) were randomised and received study agent; three patients (8.3%) discontinued study agent prior to week 22. In Part B, 151 patients were randomised and received study agent; 30 patients were randomised to each treatment group, with the exception of the sirukumab 25 mg group having 31 patients. Of these 151 patients, 20 (13.2%) discontinued study agent before week 24.

Patient disposition in (A) Part A and (B) Part B. Adverse events (AEs) leading to study agent discontinuation before week 12 were worsening of rheumatoid arthritis (placebo), cellulitis staphylococcal and pneumonia (sirukumab) in Part A; and included bacterial arthritis (placebo and sirukumab), fibrosarcoma (sirukumab), and serum sickness (sirukumab) in Part B.
Within Parts A and B, baseline demographics and disease characteristics were generally well balanced among the treatment groups, with the exceptions of the distribution of males (Parts A and B) and CRP level (Part A) (table 1). Mean patient weight in Part A was approximately 12 kg greater than that in Part B, which was likely due to differences in race (94% Caucasian vs 60% Caucasian and 21% Asian).
Baseline demographic and disease characteristics
Clinical efficacy
Part A
In Part A, patients who received sirukumab 100 mg q2w had a significantly greater mean (SD) improvement from baseline to week 12 in DAS28-CRP (2.1 (0.8) vs 0.6 (0.9), p<0.001). Sirukumab-treated patients also had a significantly greater ACR20 response rate (71.4% vs 17.6%, p=0.004), significantly greater good/moderate DAS28-CRP response rates (35.7%/57.1% vs 5.9%/23.5%, p<0.001), and significantly greater mean improvements in HAQ-DI score (0.74 vs 0.17, p<0.001) and CDAI (16.7 vs 7.2, p=0.009) (table 2). In addition, 28.6% of sirukumab-treated patients had an ACR50 response at week 12 compared with 5.9% of patients who received placebo; however, while the numerical difference between the groups was approximately fivefold, it did not reach statistical significance (p=0.148) (table 2).
Efficacy results at weeks 12 and 24 for Parts A* and B
In general, clinical response to sirukumab 100 mg q2w occurred as early as week 2. Among patients who initially received placebo, clinical response after crossover to sirukumab rapidly approached that observed among patients who initially received sirukumab. Despite sirukumab crossover to placebo at week 12, clinical response was durable and sustained through week 24 (figure 2).
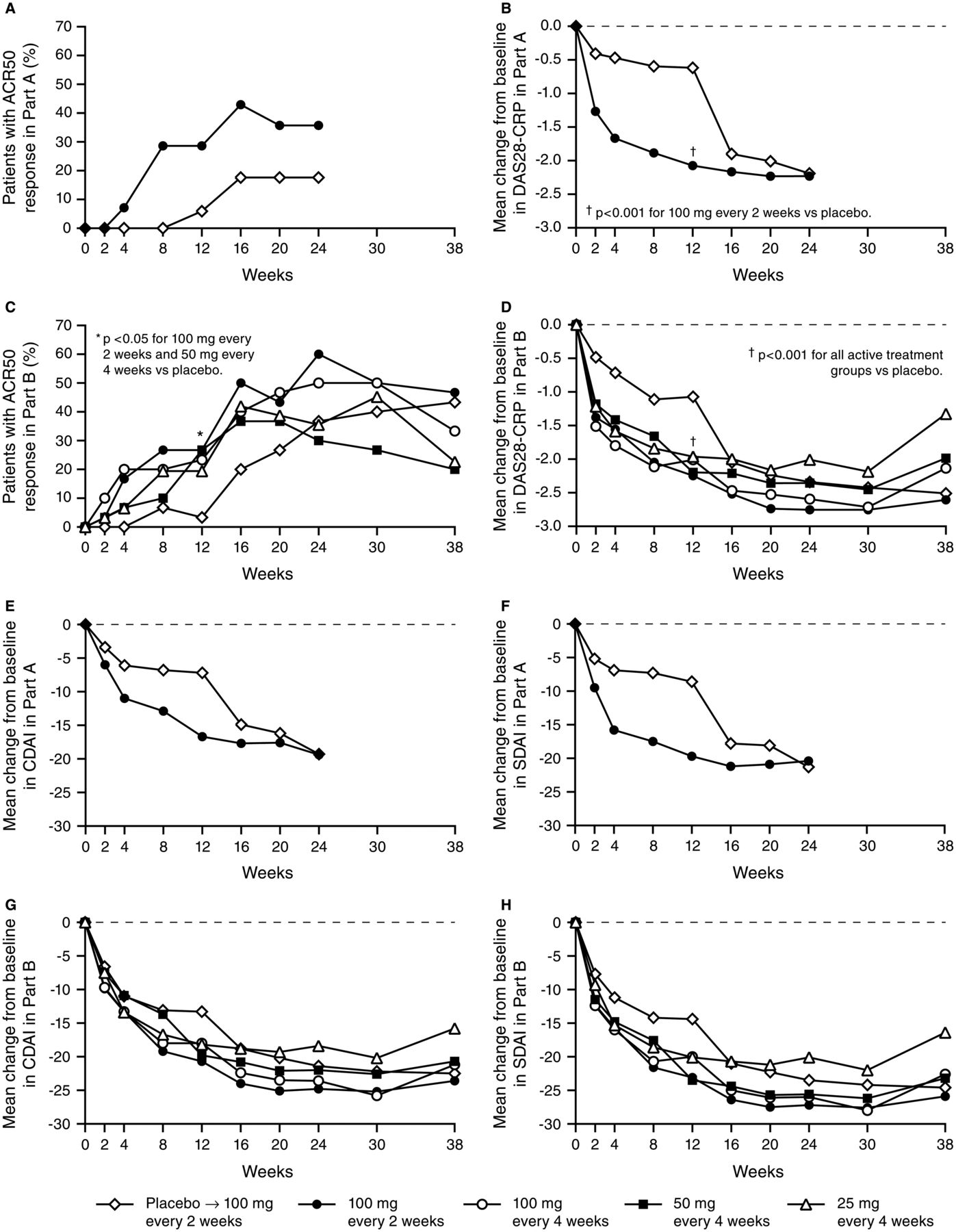
Proportions of patients with American College of Rheumatology 50 (ACR50) response (A, C) and mean changes from baseline in DAS28-C-reactive protein (CRP) (B, D), clinical disease activity index (CDAI) score (E, G), and simplified disease activity index (SDAI) score (F, H) in Parts A and B. Data from one of the study sites that participated in Part A were excluded from all efficacy analyses due to questionable data integrity. At week 12 in Part A, patients randomised to placebo crossed over to sirukumab 100 mg every 2 weeks, and patients randomised to sirukumab crossed over to placebo through week 22. At week 12 in Part B, patients randomised to placebo crossed over to sirukumab 100 mg every 2 weeks through week 24.
Part B
The primary efficacy endpoint (ACR50 at week 12) was achieved only in the sirukumab 100 mg q2w group versus placebo (26.7% vs 3.3%, p=0.026; table 2). Other sirukumab groups showed numerically greater ACR50 response rates versus placebo, although they did not achieve statistical significance. The ACR50 results were confirmed by sensitivity and subgroup analyses, and no differences were observed between geographic regions (data not shown). All four sirukumab groups had a significantly greater mean improvement in DAS28-CRP at week 12 versus placebo. Additionally at week 12, ACR20 response rates were significantly higher in the two sirukumab 100 mg groups versus placebo. Mean per cent improvements in the individual ACR core set components were numerically greater in the sirukumab groups at week 12 compared with placebo; although most differences were not significant.
Also at week 12, a greater proportion of patients in the sirukumab 100 mg q2w group achieved a good/moderate DAS28-CRP response versus the placebo group (36.7%/56.7% vs 10.0%/50.0%, p=0.003) (table 2). DAS28-CRP remission was achieved by six patients in the sirukumab 100 mg q2w group versus none in the placebo group (20.0% vs 0%, p=0.024). Overall, few patients achieved CDAI-based, SDAI-based or Boolean-based remission at week 12. Patients in the sirukumab 100 mg q2w group had a greater mean improvement in CDAI at week 12 compared with the placebo group (20.7 vs 13.3, p=0.021), in line with a major improvement in clinical variables irrespective of acute phase reactant levels. No significant differences were observed in mean improvements from baseline to week 12 in SF-36 PCS or MCS scores or HAQ-DI scores.
As in Part A, clinical response in Part B was generally rapid and often observed 2 weeks after initiating sirukumab treatment either at baseline or week 12. Efficacy was durable and was maintained for all four dose groups for at least 6 weeks after the last sirukumab administration (figure 2).
Safety
Part A
In Part A, 24 patients reported ≥1 AE (placebo: n=12, 63.2%; sirukumab: n=12, 70.6%) through week 12 (table 3). Following treatment crossover at week 12, AEs were reported by 13 patients (72.2%) receiving sirukumab, and 10 patients (62.5%) receiving placebo. Infections were the most common type of AE, specifically nasopharyngitis and upper respiratory tract infections. Two patients (sirukumab 100 mg q2w) discontinued the study due to AEs (staphylococcal cellulitis; pneumonia). Through week 38, one serious AE (SAE; staphylococcal cellulitis) occurred in a patient receiving sirukumab 100 mg q2w. No opportunistic infections, cases of tuberculosis, anaphylactic reactions, gastrointestinal perforations, or deaths occurred during Part A. Injection site reactions occurred more frequently among patients receiving sirukumab (table 3); most were considered mild.
AEs through week 38
Part B
Through week 12 in Part B, the proportions of patients with ≥1 AE were similar for the placebo and combined sirukumab groups (66.7% and 67.8%, respectively; data not shown). No dose effect was observed among the sirukumab groups (table 3). Through week 38, a total of 147 patients received sirukumab; of these, 119 (81.0%) reported ≥1 AE, with infections being the most common (n=45, 30.6%), followed by abnormal laboratory investigations (n=28, 19.0%). A total of 11 patients (placebo: n=2, 6.7%; sirukumab: n=9, 7.4%) discontinued study agent prior to week 24 due to AEs. SAEs were reported by 17 patients (placebo: n=4, 13.3%; sirukumab: n=13, 8.8%) through week 38. Among sirukumab-treated patients, SAEs included pneumonia (n=1, 100 mg q4w), increased transaminases (n=1, 50 mg q4w; alanine aminotransferase (ALT) increase grade 3 (>5.0–20.0×upper limit of normal (ULN)); aspartate aminotransferase (AST) increase grade 2 (>2.5–5.0×ULN)), and fibrosarcoma (n=1, 100 mg q4w). In Part B, one death occurred in a patient randomised to the placebo→ sirukumab group. The patient (female, aged 59 years) had a history of peripheral vascular disease and hypertension and experienced a fatal brain aneurysm at week 30.
There were no opportunistic infections, cases of tuberculosis, or gastrointestinal perforations in Part B. One patient in the 100 mg q4w group developed an AE that was reported to be serum sickness and discontinued treatment; this patient did not have any injection site reactions and tested negative for antibodies to sirukumab. As in Part A, injection site reactions in Part B were more common among patients who were receiving sirukumab (table 3); most reactions were mild.
Laboratory investigations
Haematologic changes, including decreases in white blood cells, neutrophils and platelets occurred in all sirukumab groups in Parts A and B, and generally occurred within 2 weeks of initiating treatment. Most abnormalities were classified as grade 1 or 2. In Part A, one sirukumab-treated patient had grade 3 neutropenia (<1.0–0.5×109/L). In Part B, there were three grade 3 (<0.5–0.2×109/L; one patient discontinued sirukumab) and one grade 4 (<0.2×109/L) lymphopenia; three grade 3 neutropenia (<1.0–0.5×109/L; no serious infections occurred in these patients); one grade 3 leukopoenia (<2.0–1.0×109/L); and one grade 3 (<50–25×109/L) and one grade 4 (<25×109/L) thrombocytopenia that was asymptomatic and resolved to a normal platelet count 11 days later.
In both parts, transient elevations in ALT (total n=28 with grade 2 (>2.5–5.0×ULN); total n=9 with grade 3 (>5.0–20.0×ULN)) and AST (total n=10 with grade 2 (>2.5–5.0×ULN); total n=1 with grade 3 (>5.0–20.0×ULN)), without associated increases in bilirubin or symptoms, were observed, typically within 4 weeks of sirukumab treatment with levels that declined or returned to normal without interruption of dosing.
In Part A, increases in lipid levels were observed in the sirukumab group within 2 weeks and sustained through week 12 (data not shown). After crossover to placebo, lipid levels approached baseline values by week 24. In Part B, increases in lipids were also observed within 2 weeks in the sirukumab groups and were sustained through week 24.The majority of sirukumab-treated patients in Part B with normal baseline high-density lipoprotein (HDL), low-density lipoprotein (LDL), and triglyceride levels had values that remained within normal range through week 24. However, among 78 patients with normal total cholesterol values at baseline, 56% had elevated levels at week 24. Approximately 35% of sirukumab-treated patients with normal baseline LDL levels (≤130) had values >130 at week 24 (see online supplementary table S1). These laboratory abnormalities occurred without dose relationship or short-term clinical sequelae.
Immunogenicity
In Part A, none of the 31 patients with appropriate samples (ie, ≥1 serum sample obtained after the first sirukumab dose) tested positive for antibodies to sirukumab. In Part B, two (1.4%) of 142 patients (both in the 100 mg q4w group) with appropriate samples tested positive for antibodies to sirukumab; neither patient had an injection site reaction; one patient was also positive for neutralising antibodies to sirukumab and remained an ACR20/50 responder through week 30.
Pharmacokinetics
Fourteen patients in Part A and 142 patients in Part B were included in the pharmacokinetic analyses. The mean serum sirukumab concentration-versus-time profiles are presented in figure 3. Mean half-life values were 18 days in Part A and 15−19 days across all treatment groups in Part B (see online supplementary table S2). In Part B, mean Cmax and AUC0-28d values increased in a dose-proportional manner following the first and last doses. Serum sirukumab concentrations generally achieved steady state by week 12, with mean trough concentrations of 0.99–11.63 μg/mL at week 12 for the sirukumab groups in Part B.

{kind=link}
{kind=link}
{kind=link}
Mean (SD) serum sirukumab concentration versus time after multiple subcutaneous administrations of sirukumab in (A) Part A and (B) Part B and mean per cent change from baseline in serum concentrations of C-reactive protein (CRP) in (C) Part A and (D) Part B. Data from one of the study sites that participated in Part A were excluded from all pharmacokinetics and pharmacodynamics analyses due to questionable data integrity. Serum sirukumab concentrations were plotted starting on day 2 (Part A) and day 5 (Part B).
Pharmacodynamics
Mean serum concentrations of CRP decreased significantly from baseline to week 2 in sirukumab-treated patients compared with placebo in Part A (−91% vs −20%, p<0.001) and in each sirukumab group compared with placebo in Part B (−81% to −89% vs +53%; all p<0.001). These decreases in CRP to or near the limit of detection (3.0 mg/L) were maintained through at least week 24 in Parts A and B (figure 3).
Discussion
This phase II study was the first to assess the safety and efficacy of SC sirukumab in patients with active RA despite MTX therapy. Improvement in disease activity occurred as early as 2 weeks after initiation of sirukumab treatment. The primary endpoint (ACR50 at week 12 in Part B) was achieved with sirukumab 100 mg q2w. The differences in ACR20 and ACR50 response rates between the sirukumab groups versus placebo at week 12 (27–54% and 16–23%, respectively) in this study were generally broadly consistent with those achieved with the anti-IL-6 receptor antibodies tocilizumab16 ,17 (week 16: 22–33% and 3–24%, respectively; week 24: 22–33% and 20–33%, respectively) and sarilumab18 (week 12: 3–26% and 7–25%, respectively), and the anti-IL-6 antibody clazakizumab19 (week 16: 29–46% and 26–35%, respectively) in patients with an inadequate response to prior MTX therapy. This suggests that blocking the ligand IL-6 conveys similar efficacy as interfering with the receptor for IL-6. Moreover, because the overall efficacy of IL-6R inhibition appears to be similar to that of TNF-inhibition,20 ,21 given that IL-6 and IL-6R inhibition appear to have similar efficacy by indirect comparison of phase II study results, it consequently may be speculated that monoclonal antibodies to IL-6 may convey similar efficacy as TNF-blockers. However, this assumption is not based on any head-to-head comparisons, and it should be noted that all these trials differed in their populations, definitions of MTX inadequate response, and non-allowance of DMARDs other than MTX. Moreover, currently no inhibitors of TNF receptors are approved and, therefore, it is not possible to compare the relative efficacy and safety of IL-6R and IL-6 inhibition with that of TNF-R and TNF inhibition, respectively.
Sirukumab rapidly suppressed CRP irrespective of dose level and maintained this suppression through 24 weeks. In turn, significantly greater improvements in DAS28-CRP score were observed at week 12 within all sirukumab groups when compared with placebo in Parts A and B. A few patients achieved remission at week 12; more patients achieved remission by various definitions after 24 weeks of treatment. Remission rates were highest when using the DAS28-CRP definition, which is considered to be less stringent than the CDAI, SDAI, and Boolean-based definitions.15 Importantly, the week 12 timepoint is not optimal for assessing a profound response like remission, which would be expected to occur rather at 5–6 months.22 Consistent with this expectation, remission rates for the 100 mg doses in Part B were numerically considerably higher at week 24 than at week 12.
Notably, among patients randomised to sirukumab in Part A, clinical response following crossover to placebo was maintained through week 24, despite a sirukumab mean half-life of 2.6 weeks. Additionally, in Part B, clinical response was maintained through 6 weeks after the last administration, which suggests a prolonged IL-6 inhibition.
The safety profile of sirukumab in this phase II trial was consistent with phase II–III trials of tocilizumab, sarilumab, and clazakizumab16–19 in patients with RA, with infections being a common AE for these therapies. No sirukumab dose effect was observed for AEs. No opportunistic infections, cases of tuberculosis, or gastrointestinal perforations occurred during the study. One death occurred (brain aneurysm) in the sirukumab 100 mg q2w group in Part B; whether IL-6 inhibition affects aneurysmal vessel walls is not known. Two patients (1.2%) tested positive for antibodies to sirukumab.
The effects of sirukumab on laboratory investigations were similar to those of other IL-6 pathway inhibitors.16–19 No dose response was observed for the onset or magnitude of the laboratory parameter changes. The effects of sirukumab on neutrophil and platelet counts appeared to be sustained longer with the 100 mg q2w regimen than with the other dosing regimens. This is consistent with the degree of CRP suppression over time in the various groups.
It is currently unknown if the inhibition of IL-6 rather than the IL-6R has any advantages or disadvantages. On the other hand, IL-6 can also bind to its soluble receptor and only subsequently engage the accessory molecule gp130 on the cell surface; however, IL-6R and IL-6 blockade should be able to block this soluble IL-6-sIL-6R complex and thus prevent trans-signalling from occurring. It may be for all these reasons that the indirect, rough comparisons show similar efficacy and safety of the IL-6 and the IL-6R antibodies. Nevertheless, in theory, blocking the ligand may still allow some residual activation of the receptor complex on a background or homeostatic level, while blocking the receptor completely might shut off all types of signalling mechanisms related to IL-6.
This study demonstrates proof-of-concept that IL-6 blockade by sirukumab provides a biologic therapy for RA. Improvements in the signs and symptoms of RA were observed with all sirukumab dose regimens in this trial of patients with active RA despite MTX therapy. Sirukumab rapidly suppressed CRP and maintained this suppression for at least 4 weeks after a single dose of 25, 50, or 100 mg. The results suggest that IL-6 inhibitors may have a safety profile similar to that of the IL-6R inhibitor tocilizumab in blocking the effects of IL-6. Thus, to obtain further safety insights, vigilant monitoring of the possible effects of these therapies on the cardiovascular system and hepatic, haematologic and lipid parameters is mandatory. As with any new agent, the risks of sirukumab to the individual patient should be carefully weighed against the potential treatment benefits. Furthermore, while efficacy and safety results are similar in the short term, additional data will be needed to evaluate any potential differences between therapies targeting IL-6 and those that target the IL-6 receptor among patients in RA. Phase III trials of sirukumab in RA are ongoing.
Acknowledgments
The following investigators participated in the conduct of this study: (Hungary) Regina Cseuz, Budapest; Bernadette Rojkovich, Budapest; Ferenc Szanyó, Győr; (Japan) Yukitomo Urata, Aomori; Yoshiki Shiohira, Okinawa; Yoshiya Tanaka, Kitakyushu; Koichi Amano, Saitama; Toshihiko Hidaka, Miyazaki; Yukitaka Ueki, Nagaskai; Shuji Ohta, Hitachi-City, Ibaraki; (Mexico) Fedra Irazoque, Mexico, DF; Javier Orozco, Guadalajara, Jalisco; Sergio Gutierrez, Guadalajara, Jalisco; Hilario Avila, Guadalajara, Jalisco; Ruben Burgos, Mexico, DF; (Poland) Piotr Leszczynski, Poznan; Jan Brzezicki, Elblag; Slawomir Jeka, Bydgoszcz; Artur Racewicz, Bialystok; Mariusz Korkosz, Krakow; Barbara Grabowicz, Gdynia; Anna Filipowicz-Sosnowska, Warszawa; Anna Dudek, Warszawa; Dariusz Chudzik, Lublin; (Russia) Alexander Orlov-Morozov, Moscow; Alexey Maslyanskiy, St Petersburg; Eugeny Nasonov, Moscow; Valery Shirinsky, Novosibirsk; Elena Ilivanova, St Petersburg; Tatyana Raskina, Kemerovo; (South Korea) Ho-Youn Kim, Seoul; Seung Cheol Shim, Daejeon; Young Mo Kang, Daegu; (USA) Thomas W Littlejohn, Winston Salem, NC; Amir Agha, Anderson, SC; Jane Box, Charlotte, NC; Frederick T Murphy, Duncansville, PA; Norman B Gaylis, Aventura, FL; Trial coordinators: Sheila Patil, Teresa Piperno, Patrick Gallagher (Janssen Research & Development, LLC). The authors thank Jennifer Han, MS and Rebecca Clemente, PhD, of Janssen Scientific Affairs, LLC for medical writing assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table 1
- Data supplement 2 - Online table 2
Footnotes
-
Handling editor Tore K Kvien
-
Contributors Concept/study design, data interpretation, drafting or critical revision, final approval of the manuscript, agreement to be accountable for accuracy and integrity of the work: All authors; Data acquisition: JSS, MEW, BH, YZ; Data analysis: BH, YZ, SS.
-
Funding This study was sponsored by Janssen Research & Development, LLC, a division of Johnson & Johnson. Authors BH, SS and YZ are employees of the study sponsor and were involved in study design, data collection and interpretation, drafting and revising the manuscript, and approved the final manuscript for submission.
-
Competing interests Authors YZ, SS and BH are employees of the study sponsor, Janssen Research & Development, LLC and own stock in Johnson & Johnson. JSS has received grants and/or fees from AbbVie, Astra Zeneca, BMS, Celgene, GlaxoSmithKline, Janssen, MSD, Novo Nordisk, Pfizer, Sandoz and UCB. MEW has received grants and/or fees from Ablynx, BMS, Genentech/Roche, Janssen and UCB.
-
Ethics approval Ethics Committee/IRB at each site.
-
Provenance and peer review Not commissioned; externally peer reviewed.