Article Text

Abstract
Objectives To compare 1-year clinical efficacy of (1) initial triple disease-modifying antirheumatic drug therapy (iTDT) with initial methotrexate (MTX) monotherapy (iMM) and (2) different glucocorticoid (GC) bridging therapies: oral versus a single intramuscular injection in early rheumatoid arthritis.
Methods In a single-blinded randomised clinical trial patients were randomised into three arms: (A) iTDT (methotrexate+sulfasalazine+hydroxychloroquine) with GCs intramuscularly; (B) iTDT with an oral GC tapering scheme and (C) MTX with oral GCs similar to B. Primary outcomes were (1) area under the curve (AUC) of Health Assessment Questionnaire (HAQ) and Disease Activity Score (DAS) and (2) the proportion of patients with radiographic progression.
Results 281 patients were randomly assigned to arms A (n=91), B (n=93) or C (n=97). The AUC DAS and HAQ were respectively −2.39 (95% CI −4.77 to −0.00) and −1.67 (95% CI −3.35 to 0.02) lower in patients receiving iTDT than in those receiving iMM. After 3 months, treatment failure occurred less often in the iTDT group, resulting in 40% fewer treatment intensifications. The difference in treatment intensifications between the arms required to maintain the predefined treatment goal remained over time. No differences were seen between the two GC bridging therapies. Respectively 21%, 24% and 23% of patients in arms A, B and C had radiographic progression after 1 year. Patients receiving iTDT had more adjustments of their medication owing to adverse events than those receiving iMM.
Conclusions Treatment goals are attained more quickly and maintained with fewer treatment intensifications with iTDT than with iMM. However, no difference in radiographic progression is seen. Both GC bridging therapies are equally effective and, therefore, both can be used.
Trial registration number ISRCTN26791028.
- Rheumatoid Arthritis
- DMARDs (synthetic)
- Early Rheumatoid Arthritis
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Recently EULAR updated the recommendations for the management of rheumatoid arthritis (RA).1 In patients with newly diagnosed RA the guidelines recommend that (1) the initial treatment strategy should encompass methotrexate (MTX) as monotherapy or in combination with other disease-modifying antirheumatic drugs (DMARDs), irrespective of glucocorticoids (GCs); (2) treatment is targeted to achieve remission, or low disease activity and (3) treatment should be adjusted if there is no improvement after 3 months or the target has not been reached by 6 months. When poor prognostic factors are present, biological agents should be considered if the initial treatment strategy has failed, or otherwise one could switch to a(nother) combination of DMARDs. Functional and radiological outcomes improve if current guidelines are upheld.1 ,2 Nevertheless, some major points for debate still exist.
First, 2010 criteria for RA3 are more and more often incorporated into daily practice. All current guidelines, however, were formulated using data from studies in patients fulfilling 1987 RA criteria.1 ,4 ,5 Thus trials comparing initial treatment strategies in the early phase of RA are needed for validation.
Second, several clinical trials concluded that initial combination therapy had better clinical efficacy than monotherapy; however, most rheumatologists have not implemented this in daily practice.6–10 Moreover, current guidelines do not recommend combination therapy for all patients with newly diagnosed RA.1 ,4 The principal motive for disregarding combination therapy was because (1) trials were biased by GCs, (2) patients were not DMARD naïve and (3) there were concerns about safety.11 ,12
Third, GCs have a rapid anti-inflammatory effect and are therefore used as bridging therapy to treat active disease in between initiation of DMARD(s) and the onset of their therapeutic effect.13 However, trials specifically comparing GC bridging therapies are sparse. More trials are, therefore, needed to investigate optimal dosage and tapering schemes.
Therefore, our aim was to compare in patients with very early RA the 1-year clinical efficacy of (1) initial triple DMARD therapy (iTDT) with initial MTX monotherapy (iMM), unbiased by GCs and (2) different GC bridging therapies: oral versus a single intramuscular injection.
Patients and methods
Patients
For this study, data were used from a clinical trial (ISRCTN26791028)—namely, treatment in the Rotterdam Early Arthritis Cohort (tREACH).14 tREACH, a multicentre, stratified single-blinded trial, is carried out in eight rheumatology centres in the Netherlands. Medical ethics committees at each participating centre approved the study protocol and all patients gave written informed consent before inclusion. Inclusion criteria for the tREACH are: age ≥18 years, arthritis in one or more joint(s) and symptom duration <1 year. Exclusion criteria for the tREACH are given in online supplement 1.
Patients were recruited from the outpatient clinics of all participating centres between July 2007 and April 2011. Eligible patients were stratified into three groups according to their likelihood of progressing to persistent arthritis based on the Visser model.15 For this analysis we included the high probability stratum (>70% probability of developing persistent arthritis).
Randomisation and blinding
Patients were randomised, using variable block randomisation stratified for centre, by an independent call-centre. Trained research nurses, blinded to the allocated treatment arm throughout the study, examined patients and calculated the Disease Activity Score (DAS).
Design
Patients were randomised into three groups
-
iTDT (MTX, sulfasalazine and hydroxychloroquine) with GCs intramuscularly;
-
iTDT with an oral GC tapering scheme;
-
iMM with oral GCs similar to B.
Concurrent treatment with non-steroidal anti-inflammatory drugs and intra-articular GC injections (maximum of two per 3 months) was allowed.
DMARD dosages were: MTX 25 mg/week orally (dosage reached after 3 weeks), sulfasalazine 2 g/day and hydroxychloroquine 400 mg/day (reduced to 200 mg/day after 3 months). GCs were given either intramuscularly (methylprednisolone 120 mg or triamcinolone 80 mg) or in an oral tapering scheme (weeks 1–4: 15 mg/day, weeks 5–6: 10 mg/day, weeks 7–8: 5 mg/day and weeks 9–10: 2.5 mg/day). All patients received folic acid (10 mg/week) during MTX prescription. Osteoporosis prophylaxis (risedronate 35 mg/week and calcium/vitamin D combination 500/400 mg/IU/day) was given to patients in treatment arms B and C, during the first 3 months.
A treat-to-target approach was used, aiming for a DAS <2.4.16 If DAS was ≥2.4 medication was intensified. Intensification steps were in the order (1) MTX+etanercept (50 mg/week, subcutaneously), (2) MTX+adalimumab (40 mg/2 weeks, subcutaneously) and (3) MTX+abatacept (500–1000 mg/4 weeks, intravenously, depending on weight). Treatment intensifications were the same for each treatment arm.
If DAS was <1.6 at two consecutive visits, medication was tapered. Hierarchically ordered tapering steps were: (1) biological agent, (2) sulfasalazine, (3) MTX and (4) hydroxychoroquine. Biological agent(s), MTX and sulfasalazine were gradually discontinued, whereas hydroxychloroquine was stopped immediately. A flare during tapering, defined as DAS ≥2.4, resulted in restarting full treatment, according to the stage in the protocol.
Outcomes and assessments
Patients were examined every 3 months for all outcomes, except for hand/foot radiographs, which were obtained at baseline and half-yearly.
Primary outcomes were (1) area under the curve (AUC) Health Assessment Questionnaire (HAQ) and DAS and (2) proportion of patients with radiographic progression. Secondary endpoints were disease activity (state), functional ability, EULAR response criteria,17 Boolean-defined remission criteria,18 self-assessed disease activity and medication usage over time and after 12 months of treatment.
DAS and its thresholds are used for disease state categorisations.16 Functional ability is measured with the HAQ.19 Higher HAQ scores indicate poorer function. Radiographic progression was measured with the modified Sharp–van der Heijde score (SHS).20 Radiographs were read chronologically by two out of five qualified assessors, who were blinded to the patient's identity and treatment allocation.21 Mean SHS are reported.22 Weighted κ between assessors was 0.36 with 98% agreement. The proportion of patients with radiographic progression, defined as SHS change >0.5 and >1.2 (the smallest detectable change) and >1.2 a year, was also calculated.22
EULAR response criteria are based on attained level and change in DAS (see online supplement 2).17 Boolean remission criteria are defined as having a tender joint count, swollen joint count, C-reactive protein (in mg/dL) and patient global assessment (0–10 scale) of ≤1.18 Self-assessed disease activity is measured with the RA Disease Activity Index questionnaire (RADAI).23 Higher RADAI scores indicate more active disease.
Safety monitoring and toxicity
Safety monitoring occurred according to Dutch guidelines,24 ,25 which included laboratory tests at fixed intervals. Study medication was either stopped or the dosage lowered in accordance with the protocol if (serious) adverse events14 were seen by the attending rheumatologist. MTX could be given subcutaneously if patients had gastrointestinal complaints. If MTX had to be stopped for safety reasons, leflunomide (20 mg/day) was substituted.14
Statistical analysis
Sample-size calculation was based upon AUC HAQ, using data from the BeSt study,7 where mean AUC HAQ of combination therapy and monotherapy respectively were 7.7 (SD 5.5) and 10.5 (SD 7.4). A target sample size of 270 patients per probability stratum and thus 90 patients per arm, was needed to detect the mentioned difference with a power of 80% and two-sided α=0.05. This size is sufficient to detect a difference of 6.1 AUC DAS and 20% difference in radiographic progression.14
Clinical efficacy was calculated in an modified intention-to-treat (mITT) and per-protocol analysis. In mITT analyses patients are analysed in the groups to which they were randomised, regardless of whether they received or adhered to the allocated intervention. For the primary, but not the secondary, outcomes missing values are imputed. Statistical comparison of the baseline characteristics and outcomes (after 12 months) between iTDT and iMM (arms B vs C) and both GC bridging therapies (arms A vs B) were made by Student t test, χ2 test or Wilcoxon rank-sum test, as appropriate.
We used the AUC to compare DAS and HAQ over time between treatment arms, in which missing values at each time-point were substituted with the mean value of the corresponding treatment arm. Radiographic progression was extrapolated or interpolated if the SHS was missing after 12 months.
We also performed adjusted analyses for our primary outcomes, in which we corrected for baseline imbalances, rheumatoid factor, anti-citrullinated protein/peptide antibodies and baseline HAQ, DAS or SHS, as appropriate, using multivariate analyses.
All analyses were performed for patients in the high-probability stratum and two subgroups consisting of patients with RA according to 1987 and 2010 criteria.3 ,5 All statistical analyses were carried out using STATA V.12.0. A p value <0.05 was considered statistically significant.
Results
Patients
A total of 797 patients were assessed for eligibility and of those, 568 were included. In the high-probability stratum 281 patients were randomly assigned to treatment arm A (n=91), B (n=93) or C (n=97) (figure 1). Besides an mITT analysis, we also performed a per-protocol analysis. We excluded, from our per-protocol analysis, respectively 32 (35%), 37 (40%) and 32 (33%) patients randomised to arms A, B and C (figure 1). At baseline, the symptom duration and patients fulfilling 1987 criteria for RA differed significantly between arms (table 1).
Baseline characteristics and clinical response after 12 months for each induction therapy group, according to intention-to-treat

Trial profile and protocol violations. Results are shown as number (%). 1Other reasons are: 3× no compliance, 1× pregnancy wish and 1× continuation of SASP after switch to etanercept. The figure shows the flowchart of the tREACH trial, whereas the table shows the protocol violations within the tREACH trial during the first year of follow-up. Other reasons for dropping out, in the flowchart, were incorrect randomisation and problems with communication. GCs, glucocorticoids; HCQ, hydroxychloroquine; IM, intramuscular; MTX, methotrexate; SASP, sulfasalazine; tREACH, treatment in the Rotterdam Early Arthritis Cohort.
Clinical outcome
The difference in AUC DAS between iTDT and iMM was −2.39 (95% CI −4.77 to −0.00, p=0.0497), and −0.91 (95% CI −3.17 to 1.34, p=0.42) between both GC bridging therapies. Adjusted differences were respectively −2.61 (95% CI −4.55 to −0.66, p=0.009) and −0.015 (95% CI −1.96 to 1.99, p=0.99). The largest difference in disease activity (states) between treatment arms was seen after 3 months, after which it gradually diminished (figure 2). After 12 months DAS was 0.08 (95% CI −0.34 to 0.19) lower in patients with iTDT than in those with iMM. Difference in DAS between the different GC bridging therapies was −0.20 (95% CI −0.45 to 0.04). Similar results were found in our multivariate analyses (data not shown). No differences in disease activity states were found after 12 months between iTDT and iMM, or between the two GC bridging therapies (table 2). DAS and its components over time per treatment arm are given in online supplement 4.
Clinical response for each induction therapy group, according to intention-to-treat

(Self-assessed) disease activity, functional ability and radiographic joint damage over time, stratified for induction therapy. Error bars indicate respectively 95% CIs and IQR for given means and median. DAS, Disease Activity Score; GCs, glucocorticoids; HAQ, Health Assessment Questionnaire; HCQ, hydroxychloroquine; IM, intramuscular; MTX, methotrexate; RADAI, Rheumatoid Arthritis Disease Activity Index questionnaire; SASP, sulfasalazine; SHS, modified Sharp–Van der Heijde score.
There was no significant difference in SHS after 12 months of treatment (table 2). Respectively 21%, 24% and 23% of patients in arms A, B and C had radiographic progression. The cumulative probability plots for the three treatment arms were superimposable (see online supplement 3).
Functional improvement was seen in all patients. Difference in AUC HAQ between iTDT and iMM was −1.67 (95% CI −3.35 to 0.02, p=0.052), and −0.46 (95% CI −2.04 to 1.12, p=0.57) between both GC bridging therapies (figure 2). Adjusted differences were, respectively, −1.30 (95% CI −2.45 to −0.14, p=0.028) and 0.33 (95% CI −0.84 to 1.51, p=0.58). No significant difference in functional ability was seen after 12 months (table 2). Secondary endpoints are shown in table 2 and figure 2.
We also performed a per-protocol analysis, which showed similar results (data not shown). The above-mentioned analyses were performed in both subgroups, and produced similar results (see online supplement 5 and 6).
Medication
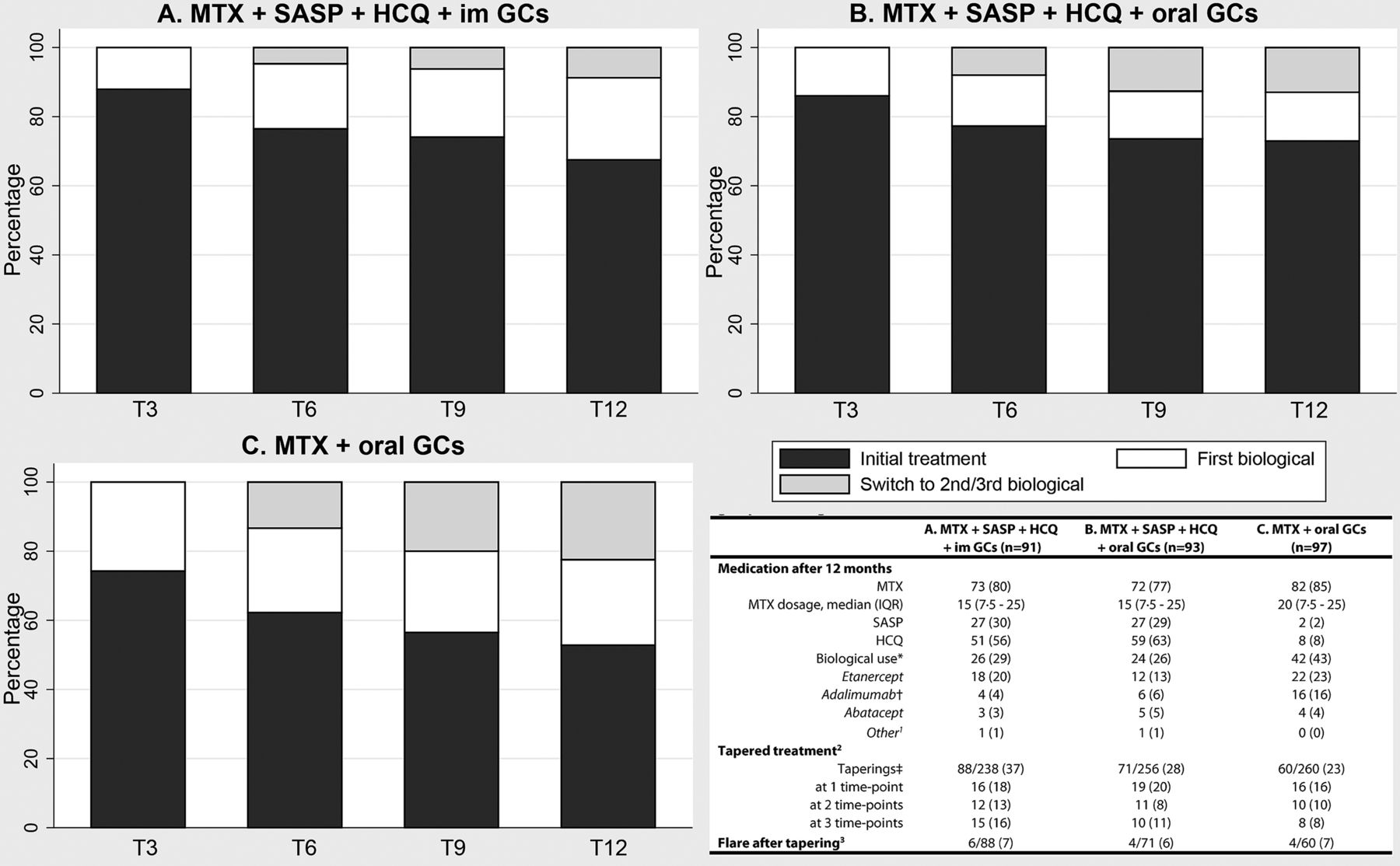
After 3 months 40% fewer biological agents were prescribed in the iTDT group than in the iMM group. This difference remained over time (figure 3). After 12 months, respectively 27% and 43% of patients, receiving iTDT and iMM, were using a biological agent (p=0.03). Moreover, for more patients receiving iMM their first biological agent had failed (16% vs 6%, p=0.03) (figure 3). In 117/281 (42%) of patients treatment could be tapered at one or more time points, and of those, 14/117 (12%) flared. Treatment could be tapered in all treatment arms, without differences in flare rates (figure 3). Biological usage did not differ between the two GC bridging therapies (figure 3). This analysis was also performed in both subgroups and showed similar results (see online supplements 5 and 6).

{kind=link}
{kind=link}
{kind=link}
Withdrawal, flares and medication usage over time and after 12 months, stratified for induction therapy. Results are shown as number (%) unless stated otherwise. 1Other biological agents are: infliximab (A) and rituximab (B). 2Treatment could be tapered after 6 months. Therefore the total amount of possible taperings is the sum of all assessments at the last three visits per treatment arm. 3A flare is defined as a Disease Activity Score ≥2.4. The proportion is calculated by dividing the number of flares by the total number of taperings. *p=0.011 for B versus C. †p=0.031 for B versus C. ‡p=0.028 for A versus B. GCs, glucocorticoids; HCQ, hydroxychloroquine; IM, intramuscular; MTX, methotrexate; SASP, sulfasalazine.
Adverse events (AEs)
No differences in serious AEs were seen (table 3). However, the proportion of patients with medication adjustments due to AEs differed significantly between iTDT and iMM (60/93 (65%) and 44/97 (45%), p=0.008). Besides switching to MTX subcutaneously, these differences vanished after stratification for drug (table 3). No differences were seen between the two GC bridging therapies. Most treatment adjustments occurred in the first 3 months (51/159, 32%). Among all patients gastrointestinal complaints and fatigue were the most commonly reported AEs, respectively 56% and 36% (table 3).
Number (%) of patients with (serious) adverse events and treatment alterations due to side effects for each induction therapy group
Discussion
In this study, unbiased for GCs, we showed that the AUC DAS was significantly less in the iTDT group than in the iMM group. A trend was observed for the AUC HAQ. Treatment goals were attained more quickly and maintained with 40% fewer treatment intensifications in the iTDT group. Moreover, for more patients receiving iMM their first biological agent had failed, reducing therapeutic options. Disease activity, functional ability and radiographic progression, after 12 months, did not differ between iTDT and iMM. No differences in serious AEs were seen. However, patients receiving iTDT had more medication adjustments due to AEs than the iMM group. Treatment could be tapered in all treatment arms, without differences in flare rates. No differences were seen between the two GC bridging therapies.
We were able to analyse the difference in efficacy between iTDT and iMM, unbiased for GCs and in DMARD-naïve patients. We found no difference in disease activity, functional ability and radiographic progression after 12 months of treatment, owing to our treat-to-target approach (intensifying treatment until the target is reached). Therefore, it is not the endpoint, but progress towards the endpoint which matters most. Aletaha et al26 have shown that the initial clinical response is related to long-term disease activity and, indirectly, the level of joint destruction and treatment changes needed to achieve the predefined treatment goals. With iTDT, treatment goals were attained more quickly, resulting in fewer treatment alterations, including the prescription of 40% fewer biological agents. However, no differences in radiographic progression were seen, which was probably owing to early initiation of intensive treatment, resulting in less joint destruction and thus less radiological progression. Moreover, the early initiation of biological agents in the iMM group might have prevented/delayed the radiographic progression.
In our trial switching to biological agents was possible after 3 months, if the target had not been reached. In the iMM group one might argue that triple DMARD therapy rather than biological agents should be the first step-up, especially since there is no evidence that step-up to a biological agent rather than triple DMARD therapy results in better clinical efficacy.10 ,27 ,28 Moreover, the updated EULAR guidelines only recommend switching to biological agents after 3 months if (1) there is no improvement in disease activity and (2) poor prognostic factors are present (ie, autoantibody positivity and erosive disease).1 Twenty of the 36 (56%) patients for whom iMM treatment had failed after 3 months were EULAR non-responders, defined as having a DAS >2.4 and <0.6 decline in DAS from baseline, with poor prognostic factors. Therefore, we think intensification to biological agents after iMM treatment had failed was reasonable.
For economic reasons efficient use of biological agents is needed to be able to continue optimal rheumatic care in the future.29 With iTDT treatment, goals are attained more quickly and maintained with 40% fewer biological agents, reducing costs enormously. Better disease control improves worker productivity, and thus costs due to loss of productivity are also reduced.30 ,31 However, the cost–utility analysis of the tREACH trial still has to reconfirm this statement.
Patients and/or rheumatologists, however, may be averse to iTDT treatment, mainly because of the large amount of drugs that have to be taken. Medication adherence in RA is strongly influenced by a patient's belief about the need for the drugs.32 These beliefs are moulded by rheumatologists through the information given about the disease and treatment approach.32 A personalised medicine approach would be ideal in this very early phase, especially since 60% respond well to iMM; conversely, therefore, some patients receiving iTDT are overtreated. Determination of early GC response after treatment initiation is a promising predictor, possibly leading to a more personalised medicine approach.33 Therefore, we think that future research should focus on developing a more personalised treatment approach, in which differentiation between patients who would thrive on iMM and those who need iTDT might be a first step.
In 42% of patients treatment could be tapered, and of those, 12% flared. Therefore, we think tapering DMARDs and/or biological agents is justified in patients with sustained remission. However, patients should still be monitored strictly during tapering. Data on tapering medication are sparse, especially in early RA.1 ,34 Future research is needed to determine (1) when to start tapering, (2) how to taper and (3) the optimal interval between taperings.
We found that intramuscular and oral GCs are equally effective as bridging therapy, but one single injection might be more feasible. However, duration of our GC tapering scheme was short (10 weeks) and the initial dosage was low (15 mg) in comparison with, for example, the COBRA regimen (respectively 28 weeks and 60 mg).9 Because GCs have disease-modifying traits with longlasting benefits even after withdrawal, a different GC oral tapering scheme might be superior.13 However, if we compare our iMM treatment with the COBRA-light strategy, intensification to biological agents after 6 months is equally indicated (respectively 36% and 41%).35 In the COBRA-light trial, however, the treatment goal is remission instead of low disease activity, which prevents useful direct comparison.35 Therefore, future research is needed for optimising GC bridging therapies.
Our study had certain limitations. Foremost, baseline imbalances occurred, despite randomisation, which is why we also performed an adjusted analysis. After adjustment similar results were found for the primary outcomes, but the difference in AUC HAQ became significant (favouring iTDT). Additionally, only research nurses, who assessed the DAS, were blinded to the allocated treatment arm. This design was chosen, since we wanted to mimic daily practice as far as possible. Single blinding, however, might be a potential source of bias, because of the aversion for iTDT by rheumatologists and/or patients (favouring iMM) or (un)intentional misinformation due to the rheumatologist's beliefs (favouring iTDT). Moreover, for various reasons 101 (36%) patients were excluded from our per-protocol analysis, which was more than expected. Our exclusion percentage, however, was comparable with that of other trials.9–10 ,27
In conclusion, in our treat-to-target design, treatment goals were attained more quickly and maintained with fewer treatment intensifications, with iTDT than with iMM. However, no difference was seen in radiographic progression. Before choosing the initial treatment strategy, rheumatologists should be aware of the benefits and risks, but additionally, known prognostic factors and the patient's wish should be taken into account. One single intramuscular GC injection and a low-dose oral GC tapering scheme would be sufficient as bridging therapy.
Acknowledgments
We thank all patients who are enrolled in the tREACH trial. Without their active cooperation, our trial would not be possible. The tREACH trial comprises the following rheumatology centres: Erasmus MC, Rotterdam; Sint Franciscus Gasthuis, Rotterdam; Maasstad Ziekenhuis, Rotterdam; Vlietland Ziekenhuis, Schiedam; Admiraal de Ruyter Ziekenhuis, Goes and Vlissingen; Zorgsaam Ziekenhuis, Terneuzen; Albert Schweitzer Ziekenhuis, Dordrecht. We thank the following people (in alphabetical order) from all centres, for their contribution to the tREACH trial: Aartsen R, Alfenaar C, Alves C, Arendse R, Arnoldus M, Baak-Dijkstra M, Bal-overzier J, Barendregt P, Basoski N, Beer S, Berkel D, Bonte F, Born van den M, Breukelen van D, Bron S, Buijs H, Buijs N, Cambier M, Cate ten D, Claessen S, Colin E, Dekker A, Dolhain R, Donze M, Fodili F, Gerards A, Goudeketting G, Grillet de B, Haasnoot H, Hamelink B, Han K, Houdt van Y, Hove van L, Huisman M, Jager de J, Jager de M, Jasperse J, Jonkers C, Joziasse S, Kaal-Alfenaar C, Klootwijk K, Krommenhoek T, Krugten van M, Kuijper T, Lam Tse W, Leemput van H, Legierse C, Lenssinck M, Lubbe van de P, Maclean P, Man de Y, Matena D, Molenaar A, Mous L, Nijs J, Paassen van H, Quax R, Reijnierse J, Romme A, Rotte de M, Saltzherr M, Schaeybroeck B, Schardijn G, Schrauwen S, Sonnaville de P, Steenwinkel de F, Sturm L, Sutter T, Tchetverikov I, Tusschenbroek D, Veldman R, Vis M, Voordt van der A, Voorneveld H, Vroed de M, Walravens M, Walter M, Werff van de N, Westeinde van het A, Wiele J, Willemse M, Wouter J, Zandbergen W, Zeben van D, Zwart H.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Handling editor Tore K Kvien
-
Contributors All authors contributed to the conception or design of the trial; or the acquisition, analysis, or interpretation of data for the work; drafting or revision of the manuscript; and final approval of the manuscript for publication.
-
Funding This work was supported by an unrestricted grant from Pfizer bv. [0881–102217]. Pfizer had no involvement in the study design; in collection, analysis and interpretation of data; writing of the report; and decision to submit for publication. The corresponding author had full access to all data and had final responsibility for the decision to submit for publication. Data management was sponsored by the Dutch Arthritis Foundation.
-
Competing interests None.
-
Ethics approval Ethics committee of the Erasmus MC.
-
Provenance and peer review Not commissioned; externally peer reviewed.