Article Text

Abstract
Objective Report on radiographic effects and maintenance of clinical benefit with intravenous golimumab 2 mg/kg+methotrexate (MTX) for up to week (wk) 52 in active rheumatoid arthritis (RA).
Methods Patients (n=592) with active RA (≥6/66 swollen, ≥6/68 tender joints, C reactive protein (CRP) ≥1.0 mg/dL and positive for rheumatoid factor and/or anticyclic citrullinated protein antibody at screening) despite MTX ≥3 months (stable dose of 15–25 mg/week for ≥4 weeks) participated in this multicentre, international, randomised, double blind, placebo controlled, phase 3 study. Patients were randomised (2:1) to receive intravenous golimumab 2 mg/kg or placebo infusions at weeks 0 and 4 and then every 8 weeks; patients continued their stable MTX regimen. Placebo patients started golimumab 2 mg/kg at wk16 (early escape; <10% improvement in tender and swollen joints) or wk24 (crossover by design). Week 24 and wk52 radiographic (van der Heijde-Sharp (vdH-S) scores), clinical efficacy and safety data up to 1 year are reported here.
Results Significant and rapid clinical improvement was observed up to wk24 of intravenous golimumab therapy. Golimumab+MTX treated patients demonstrated less radiographic progression than placebo treated patients at wk24 (vdH-S score mean change 0.03 vs 1.09; p<0.001) and wk52 (0.13 vs 1.22; p=0.001). Among patients with ≥20% improvement in the American College of Rheumatology response criteria or who achieved a ‘good’ or ‘moderate’ response according to the 28 joint Disease Activity Score employing CRP at wk24, approximately 80% maintained this response up until wk52. Through an average of 43.5 weeks of follow-up, 64.6% of patients receiving golimumab+MTX reported adverse events, most commonly non-serious infections.
Conclusions In patients with active RA despite MTX, intravenous golimumab+MTX yielded significant inhibition of structural damage at wk24 and wk52, and sustained clinical improvement in signs and symptoms with no new safety signals up to 1 year.
ClinicalTrials.gov NCT00973479, EudraCT 2008–006 064–11.
- Rheumatoid Arthritis
- TNF-alpha
- Methotrexate
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Patients with rheumatoid arthritis (RA) who demonstrate an inadequate response to disease modifying antirheumatic drugs, such as methotrexate (MTX), are often treated with one or more antitumour necrosis factor (TNF) agents.1 The human monoclonal antibody, golimumab, has demonstrated safety and efficacy in phase 3 trials in the treatment of patients with RA when administered via subcutaneous injection,2–4 as well as when administered as an intravenous infusion.5 ,6
In the recent GO-FURTHER trial, intravenous golimumab was given as a 2 mg/kg infusion at week 0 (wk0) and wk4, followed by every 8 week (q8w) maintenance therapy.6 Six month results indicated that the addition of intravenous golimumab rapidly and significantly improved RA signs and symptoms, and physical function in patients with active RA, despite ongoing MTX therapy.6 We now report radiographic findings at wk24 and wk52 and clinical efficacy and safety findings assessed after up to 1 year of the GO-FURTHER trial.
Patients and methods
Patients
The GO-FURTHER patient eligibility criteria have been detailed previously (also see online supplementary text).6 Adults with active RA despite MTX for ≥3 months (stable regimen of 15–25 mg/week for ≥4 weeks) were enrolled. Active RA was defined by ≥6 of 66 swollen joints and ≥6 of 68 tender joints at screening and baseline, and a screening C reactive protein (CRP) concentration ≥1.0 mg/dL (upper limit of normal 1.0 mg/dL). All patients were positive for rheumatoid factor and/or anticyclic citrullinated protein at screening and naïve to prior anti-TNF treatment.
Study design and assessments
Patients enrolled into the multicentre, double blind, placebo controlled GO-FURTHER study were randomly (2:1) assigned, via an interactive voice response system, and stratified by screening CRP (<1.5 mg/dL, ≥1.5 mg/dL) and investigational site, to receive intravenous golimumab 2 mg/kg or placebo infusions at wk0, wk4 and then q8w up to wk100, followed by 12 weeks of safety follow-up (also see online supplementary text). Patients randomised to placebo with <10% improvement in swollen and tender joint counts from baseline to wk16 early escaped (EE'd) from placebo to intravenous golimumab 2 mg/kg, beginning at wk16 and wk20, followed by q8w maintenance infusions. Placebo patients who did not EE crossed over to golimumab 2 mg/kg at wk24 and wk28 and then q8w. Patients assigned to golimumab also received placebo infusions at wk16 and wk24 to maintain blinding, regardless of EE status; escalation beyond 2 mg/kg was not permitted. All patients continued to receive a stable regimen of MTX (≥15 but ≤25 mg/week).
Radiographs of the hands and feet obtained at baseline, wk24 and wk52 or on study agent discontinuation (unless obtained within the previous 8 weeks) were assessed using van der Heijde-Sharp (vdH-S) scoring.7 In both randomised groups, patients with <10% improvement in joint counts had radiographs of the hands and feet obtained at wk16 rather than at wk24 and the wk24 scores were determined via linear extrapolation using baseline and wk16 scores. Radiographic images were evaluated by two independent readers (and an adjudicator in predefined cases) blinded to time point, patient identity and treatment (also see online supplementary text).
The study's primary endpoint was the proportion of patients achieving ≥20% improvement in the American College of Rheumatology (ACR20) response criteria8 at wk14 (reported previously).6 Four major secondary endpoints included: wk14 28 joint Disease Activity Score employing CRP (DAS28–CRP) response (European League Against Rheumatism good or moderate response), wk14 change from baseline in the Health Assessment Questionnaire-Disability Index (HAQ-DI), at least 50% improvement in ACR (ACR50) response criteria at wk24 (all reported previously6) and wk24 change from baseline in vdH-S score (reported herein).
Proportions of patients achieving a DAS28–CRP <2.69–12 were also determined. Improvement in functional status was assessed using the HAQ-DI.13 An improvement in HAQ-DI ≥0.25 units was considered clinically significant.14 Simplified Disease Activity Index (SDAI) and Clinical Disease Activity Index (CDAI)15–17 scores up to wk52 were also determined in a post hoc analysis. Results of quality of life assessments up to wk52 will be reported separately. Blood samples were evaluated for determination of serum golimumab concentrations and antibodies to golimumab using the respective validated immunoassays.18 Safety endpoints assessed up to wk52 included the frequency and type of adverse events (AEs) and serious AEs, and changes from baseline in clinical laboratory parameters.
Statistical analyses
Patient data were summarised using descriptive statistics (also see online supplementary text). Statistical analyses were conducted by Janssen Research & Development and reviewed by all of the authors. Hypothesis testing in the primary and major secondary analyses was controlled by sequential testing to adjust for multiplicity. All statistical tests were performed at a two sided significance level of 0.05.
In the primary and major secondary efficacy analyses, the proportions of patients achieving a response were compared between treatment groups using a two sided Cochran–Mantel–Haenszel test (for dichotomous endpoints) and analysis of variance on the van der Waerden19 normal scores (for continuous endpoints), stratified by screening CRP (<1.5, ≥1.5 mg/dL). Additional details of clinical efficacy analyses for data collected at visits scheduled up to wk24 have been reported previously.6 For wk52 clinical efficacy data, intent to treat analyses were performed, whereby patients were assessed by their initially randomised treatment group; patients who EE'd or crossed over to active treatment remained within their original randomised group. Last observation carried forward methodology was employed for imputation of missing data for continuous variables. Analyses of radiographic data were conducted according to the patient’s randomised treatment group. Because patients who EE'd had a radiograph at wk16 rather than wk24, the wk24 vdH-S score of these patients was determined via linear extrapolation using baseline and wk16 scores.
Results
Patient disposition and baseline characteristics
The first patient was screened on 14 September 2009, and the final patient enrolled received the wk52 infusion on 25 November 2011. The study was conducted at 92 sites in three Latin American (119 patients), five European (355 patients), one North American (61 patients) and four Asia Pacific (57 patients) countries. The wk52 database was locked on 1 March 2012 to include additional safety data. Among the 592 randomised patients, 570 (96.3%) completed the 24 week study period and 22 (3.7%) discontinued study agent before wk24.6 An additional 20 patients discontinued the study agent from wk24 to wk52, most due to AEs (seven patients) or withdrawal of consent (five patients). Additional patient disposition information is provided in the online supplementary figure S1.
Baseline patient and disease characteristics indicative of active disease have been reported previously.6 The largely Caucasian (80.4%) and female (81.6%) population entered the study with an overall median disease duration of 4.7 years (also see table 1 for additional baseline data).
Summary of demographics and disease characteristics at baseline (randomised patients)
Radiographic findings
Radiographic progression up to week 24
Patients who received golimumab 2 mg/kg+MTX demonstrated significantly less progression from baseline to wk24 in total vdH-S score versus patients receiving placebo+MTX (mean changes 0.03 vs 1.09; p<0.001) (table 2, figure 1A) (see also online supplementary text). Similar results were observed in the prespecified analysis for patients with screening CRP ≥1.5 mg/dL or <1.5 mg/dL (table 2). Additional sensitivity analyses conducted (see online supplementary text—statistical analyses) for this major secondary endpoint were confirmatory—that is, they yielded statistically significant treatment differences and demonstrated little difference from that based on the primary analysis (data not shown). In addition, the results of subgroup analyses based on baseline demographics (see online supplementary figure S2C) supported the efficacy of golimumab+MTX in inhibiting radiographic progression. As part of these subgroup analyses, patients with CRP <1.0 mg/dL and CRP >1.0 mg/dL at wk0 who received golimumab+MTX both demonstrated statistically significant treatment differences in radiographic progression compared with placebo+MTX treated patients (data not shown).
Summary of change from baseline to week 24 in van der Heijde-Sharp score (randomised patients)

Changes in van der Heijde-Sharp (vdH-S) scores at week 24: total, erosion and joint space narrowing scores (A). Cumulative probability plots for total vdH-S scores (B). Patients with change of 0 or less in the total vdH-S score (C) and patients with change in excess of the smallest detectable change (SDC) (D). GLM, golimumab; MTX, methotrexate; PBO, placebo.
The cumulative probability plot for total vdH-S scores (figure 1B) demonstrated clear separation, with greater inhibition of progression and fewer numbers of patients with progression among golimumab than placebo treated patients. Larger proportions of patients who received golimumab+MTX than placebo+MTX demonstrated a change of 0 or less in vdH-S score (70.6% vs 57.4%, respectively; p=0.001) (figure 1C). Similarly, lower proportions of golimumab+MTX than placebo+MTX treated patients demonstrated radiographic progression based on the smallest detectable change (SDC)20 in overall radiographic progression (SDC=1.91; 8.6% vs 19.3%, respectively; p<0.001), in erosion alone (SDC=1.57; 3.8% vs 10.7%; p=0.001) and in joint space narrowing (JSN) alone (SDC=1.19; 9.9% vs 17.3%; p=0.010) (figure 1D).
Significant improvements with golimumab+MTX versus placebo+MTX were observed in erosion (mean changes of −0.12 vs 0.53, respectively; p<0.001) and JSN (0.14 vs 0.56, respectively; p=0.002) scores (figure 1A). The cumulative probability plots for erosion and JSN scores (see online supplementary figures S2A and S2B) demonstrated clear separation, with greater inhibition of progression and fewer numbers of patients with progression among golimumab than placebo treated patients.
Radiographic progression up to week 52
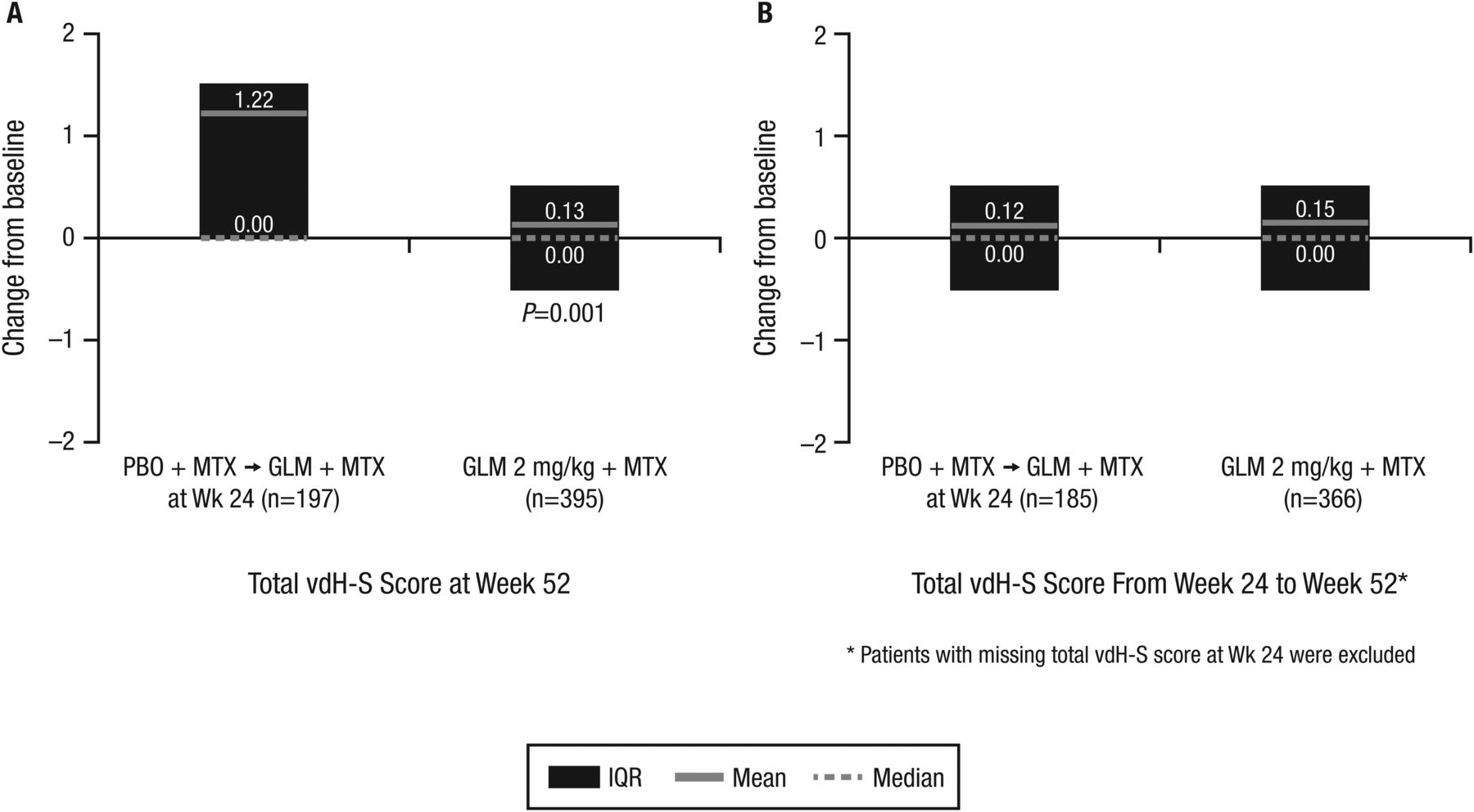
At wk52, patients randomised to placebo+MTX (who received golimumab+MTX from wk16 to wk52 or from wk24 to wk52) exhibited significantly more radiographic progression than those receiving golimumab+MTX beginning at wk0 (1.22 vs 0.13; p=0.001) (figure 2A). Between wk24 and wk52, patients initially randomised to golimumab+MTX generally demonstrated minimal change in total vdH-S score (mean change of 0.15). Importantly, patients initially randomised to placebo+MTX who began golimumab at wk16 or wk24 also demonstrated limited radiographic progression between wk24 and wk52 (mean change of 0.12), with progression being similar to patients initially randomised to golimumab+MTX (figure 2B).
{kind=link}
{kind=link}
Changes in total van der Heijde-Sharp (vdH-S) scores from baseline to week 52 (A) and from week 24 to week 52 (B). GLM, golimumab; MTX, methotrexate; PBO, placebo.
Clinical efficacy assessments
As reported previously, the study's primary endpoint (ie, ACR20 response at wk14) was achieved (p<0.001).6 A capsular summary for the primary and three of the four major secondary endpoints is provided in table 3.
Summary of clinical efficacy (randomised patients)
At wk52, ACR20, ACR50 and at least 70% ACR improvement (ACR70) response rates were 65.8%, 38.7% and 18.2%, respectively, among patients who received golimumab+MTX for a full year. Importantly, 82% (201/245) of these patients who achieved ACR20 responses by wk24 maintained these responses at wk52, and 40.1% (55/137) of ACR20 non-responders at wk24 subsequently achieved an ACR20 response by wk52. Similarly, 71.7% and 60.9% of ACR50 and ACR70 responders, respectively, at wk24 maintained these levels of response at wk52 (table 3).
Also at wk52, the DAS28–CRP response (eg, good/moderate) rate was maintained at 81.3% among patients initially randomised to golimumab+MTX. Importantly, 88.0% of patients who achieved a DAS28–CRP good/moderate response at wk24 maintained this level of efficacy up to wk52 (table 3; see also online supplementary figure S3).
By wk52, all golimumab treated patients demonstrated mean improvements from baseline in CDAI of 22 points and in SDAI of 23 points. In addition, while the vast majority of patients demonstrated high disease activity at baseline, approximately 75% of patients who initiated golimumab+MTX reached remission or low/moderate disease activity by these indices at wk52 (table 3).
Similarly, the early improvements seen in HAQ-DI scores during the initial study period were generally maintained among patients randomised to golimumab+MTX, with median and mean changes from baseline to wk52 of 0.38 and 0.51, respectively, in comparison with median and mean changes from baseline to wk14 of 0.50 and 0.50, respectively.6
Golimumab pharmacokinetics and immunogenicity
Golimumab concentrations reached steady state at wk12, with median trough serum golimumab concentrations of 0.27, 0.21 and 0.30 μg/mL at wk12, wk20 and wk52, respectively, suggesting maintenance of drug exposure up to wk52 with q8w administration of golimumab 2 mg/kg. The overall cumulative incidences of antibodies to golimumab at wk24 (3.0%, 13/440) and wk52 (4.6%, 26/560) were low. In all 26 antibody positive patients up to wk52, antibodies were neutralising to study agent. Serum golimumab concentrations were generally lower in patients who tested positive for antibodies to golimumab than in patients without evidence of antibodies to golimumab (data not shown). Overall, at wk52, ACR20 and ACR50 responses were observed in 14 (53.8%) of 26 and five (19.2%) of 26 patients who were antibody positive, respectively, versus 353 (66.1%) of 534 and 203 (38.0%) of 534 patients who were antibody negative, respectively. While no definitive conclusions regarding the relationship between positive antibody status and efficacy can be made due to the small number of antibody positive patients, these data suggest that anti-golimumab antibodies may diminish the agent's clinical efficacy. No relationship between antibodies to golimumab and the development of infusion reactions was evident, with 3.6% of all patients, 4.6% of antibody positive patients and 4.3% of antibody negative patients having had such reactions (also see ‘Safety’ below).
Safety
Up to wk52, 64.6% of all golimumab+MTX treated patients reported AEs, reflecting an increase from the 52.9% AE rate reported up to wk24.6 The most commonly reported AEs were upper respiratory tract infection, bronchitis, headache, nasopharyngitis, hypertension, pharyngitis and urinary tract infections (see online supplementary table S1). Similarly, the rate of serious AEs among all golimumab+MTX treated patients increased from wk24 (4.1%)6 to wk52 (8.6%). Serious infections continued to comprise the most commonly reported serious AEs, occurring in 1.9% of all golimumab+MTX treated patients (see online supplementary table S1); infection types appeared to be predominantly singular events. One patient in Mexico, whose baseline skin and serum tuberculosis (TB) testing was negative, was diagnosed, approximately 6 months after symptom onset, with fatal abdominal TB. No serious opportunistic infections were documented up to wk52; one patient in South Korea developed uncomplicated cryptococcal pneumonia after wk52. The patient recovered following one dose of intravenous fluconazole 100 mg followed by 6 months of oral fluconazole 400 mg once daily.
Two additional patients enrolled in the study died between wk24 and wk52. One patient in the placebo+MTX group died prior to wk24 due to a presumed stroke resulting from hypertensive crisis.6 A 32-year-old woman in the golimumab+MTX group died at approximately wk36 from a presumed myocardial infarction (no autopsy was performed) secondary to community acquired pneumonia.
Among all golimumab+MTX treated patients, three patients were diagnosed with a malignancy (a previously reported case of breast cancer prior to wk24,6 one case of cervical carcinoma stage 0 and a basal cell carcinoma between wk24 and wk52). No lymphomas or leukaemias were reported up to 52 weeks. One case of flare of pre-existing chronic lymphocytic leukaemia, Rai stage 1, occurred after the wk52 database lock in a 55-year-old patient with an extensive family history of chronic lymphocytic leukaemia. No cases of demyelination were reported up to wk52.
Three serious cardiovascular events occurred between wk24 and wk52, including unstable angina (placebo→golimumab+MTX), atrial fibrillation (golimumab+MTX) and myocardial infarction (golimumab+MTX; discussed above). No new onset of congestive heart failure was observed up to wk52.
The rate of infusion reactions (0.7%) and the proportions of patients with infusion reactions (3.6% overall, 4.6% antibody positive, 4.3% antibody negative), most commonly skin reactions (six patients with rash reactions), cardiac (four patients, bradycardia/palpitations) and vascular (four patients, hypotension/hypertension/flushing) disorders, in the golimumab+MTX group continued to be low up to wk52 (see also online supplementary table S1). No infusion reaction was serious or severe, regardless of antibody to golimumab status, and no delayed hypersensitivity or serum sickness-like reactions were observed. One patient (golimumab+MTX) had an anaphylactic reaction caused by anaesthesia for a minor surgical procedure (bronchoscopy).
Discussion
In the multicentre, double blind, placebo controlled GO-FURTHER study, patients with active RA despite background MTX therapy received intravenous golimumab 2 mg/kg or placebo infusions, administered at wk0 and wk4 followed by q8w dosing, along with stable background MTX. As previously reported, addition of intravenous golimumab 2 mg/kg to MTX rapidly and significantly improved RA signs and symptoms and was generally well tolerated by patients with active RA despite ongoing MTX therapy.6 Radiographic findings now reported indicate that patients receiving golimumab 2 mg/kg+MTX demonstrated significantly less progression from baseline to wk24 in total vdH-S, erosion and JSN radiographic scores compared with patients treated with placebo+MTX. Between wk24 and wk52, patients initially randomised to golimumab+MTX demonstrated minimal change in the total vdH-S score. Importantly, patients initially randomised to placebo+MTX who began golimumab at wk16 and wk24 also demonstrated limited radiographic progression between wk24 and wk52, with progression similar to patients initially randomised to golimumab+MTX. These findings appear to demonstrate stabilisation of radiographic disease within 6 months after the onset of golimumab+MTX therapy.
Patients enrolled in the GO-FURTHER trial of intravenous golimumab appear to have had more active inflammation (baseline median CRP of 1.9 mg/dL) and more radiographic progression (baseline mean total vdH-S score of 48.5) at study outset versus patients with RA assessed in the GO-FORWARD trial of subcutaneous golimumab (CRP of 0.8–1.0 mg/dL and (baseline mean total vdH-S scores of 29.7–39.6),21 which may be one of the reasons for differences in radiographic progression between studies. Results may have also been influenced by differential drug exposure/serum concentrations, although this hypothesis does not explain marked differences in placebo progression rates.
The longer term clinical efficacy and safety data also reported here showed sustained clinical improvement and a stable safety profile with 1 year of intravenous golimumab+MTX therapy. Importantly, more than 80% of golimumab patients who achieved ACR20, DAS28–CRP (good/moderate) and/or HAQ-DI (improvement ≥0.25 units) responses by wk24 maintained these responses at wk52. In addition, patients whose golimumab therapy was delayed by 4–6 months achieved response rates at 1 year that matched those of patients starting golimumab at study outset.
Intravenously administered golimumab continued to be well tolerated by study participants during the second half of the first year of therapy, with no new safety signals identified from the safety assessments made at wk24.6 Serious infections continued to comprise the most commonly reported serious AE. The single patient who died from abdominal TB, despite a rigorous TB screening protocol at study entry,22 and the patient who developed (and recovered from) uncomplicated cryptococcal pneumonia after wk52 underscore the need for continued vigilance in screening and following patients treated with anti-TNF agents, as well as other biologic therapies that impact on patient immune function.23 As was observed with subcutaneous golimumab in patients with RA up to wk52,24 few additional patients developed antibodies to golimumab after wk24. The presence of antibodies did not appear to influence the occurrence of infusion reactions, similar to the pattern observed with subcutaneous golimumab and injection site reactions.
Intravenous golimumab safety findings, including the occurrence of infusion reactions and serious infections, reported here were consistent with those observed in patients with active RA despite prior MTX therapy for whom subcutaneous dosing was the sole route of golimumab administration.24 The comparable nature of intravenous versus subcutaneous safety findings are also supported by data from an intravenous to subcutaneous switch golimumab trial of patients with RA, in which AE rates adjusted for length of follow-up were generally comparable between intravenous and subcutaneous golimumab admin-istration.25
In summary, intravenously administered golimumab appears to maintain clinical effect and significantly inhibits radiographic progression up to 1 year of treatment. Continued review of data to wk112 is planned.
Acknowledgments
The authors thank Michelle Perate, MS, a paid consultant for Janssen Services, LLC, for writing assistance. The authors also thank the GO-FURTHER study investigators.6
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figure 1
- Data supplement 3 - Online figure 2
- Data supplement 4 - Online figure 3
Footnotes
-
Handling editor Tore K Kvien
-
Contributors MEW, RW, AMM, DB, COB – study design and conduct, data interpretation, manuscript preparation, and decision to submit for publication. LK, KHL, SS, LN, JL, ZX, JLeu – data analysis, data interpretation, manuscript preparation and decision to submit for publication.
-
Funding Janssen Research & Development, LLC and Merck/Schering-Plough provided support for this study.
-
Competing interests COB, MEW and RW have received consulting fees from Janssen. AMM, LK, KHL, SS, LN, JL, ZX, JLeu and DB are employees of Janssen.
-
Ethics approval The protocol was approved by each site's institutional review board or ethics committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.