Article Text

Abstract
Objectives BUILDER-1 and BUILDER-2 aimed to assess the efficacy and safety of tocilizumab (TCZ) in patients with ankylosing spondylitis (AS).
Methods BUILDER-1 was a two part, phase II–III parallel-group trial in patients with AS naive to antitumour necrosis factor (aTNF) treatment. Patients in part 1 received TCZ 8 mg/kg or placebo for 12 weeks. In part 2 (beginning after part 1 enrolment ended), newly enrolled patients received TCZ 4 or 8 mg/kg or placebo for 24 weeks. The same treatment arms were used in BUILDER-2, a phase III study in aTNF-inadequate responders. The primary endpoint for both studies was the proportion of patients achieving 20% improvement in the Assessments in Axial SpondyloArthritis international Society (ASAS). Secondary and exploratory endpoints included ASAS40 response rates, Bath Ankylosing Spondylitis Disease Activity Index improvement, changes in joint counts, enthesitis score and C reactive protein (CRP).
Results 102 patients were randomised in BUILDER-1 part 1; 99 (48 TCZ, 51 placebo) completed 12 weeks. Week 12 ASAS20 response rates were 37.3% and 27.5% in the TCZ and placebo arms, respectively (p=0.2823). Secondary and exploratory endpoints did not differ between treatment arms. CRP levels declined with TCZ treatment, suggesting adequate IL-6 receptor blockade. As a result, BUILDER-1 part 2 and BUILDER-2 were terminated. TCZ safety results were consistent with previous observations in rheumatoid arthritis, except for a cluster of anaphylactic and hypersensitivity events at Bulgarian study sites. No apparent explanation for this clustering could be found.
Conclusions BUILDER-1 failed to demonstrate TCZ efficacy in treating aTNF-naive patients with AS.
Clinical trial registration numbers: NCT01209702 and NCT01209689 (www.clinicaltrials.gov).
- Ankylosing Spondylitis
- Treatment
- Inflammation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Ankylosing spondylitis (AS; also referred to as axial radiographic spondyloarthritis) is a chronic, debilitating, gradually progressive inflammatory rheumatic disease that primarily affects the axial skeleton and sacroiliac joints but also affects the peripheral joints and entheses.1 Extra-skeletal inflammation may involve the eyes, skin, cardiovascular system and gastrointestinal tract.2 AS most commonly affects men younger than 45 years of age, has a peak onset in the second and third decades of life and has a worldwide prevalence of approximately 0.5%.3
Non-steroidal anti-inflammatory drugs (NSAIDs), including selective cyclo-oxygenase 2 inhibitors, are the recommended first-line pharmacological therapy for AS, but many patients achieve insufficient symptom control with NSAIDs alone.4 ,5 Conventional disease-modifying antirheumatic drugs (DMARDs) are of limited use.4 ,6–8 Sulfasalazine and methotrexate are not effective for the treatment of patients with axial disease.4 ,6–8 Sulfasalazine and possibly methotrexate may be helpful in treating patients with peripheral arthritis.4 ,8
Antitumour necrosis factor (aTNF) therapy (eg, infliximab, adalimumab, etanercept, golimumab) is recommended for patients with persistently high disease activity despite NSAID and non-pharmacological therapy.4 Although aTNF agents significantly improve clinical symptoms in patients with AS,9–16 approximately 40% of patients do not respond adequately, and there is no evidence of inhibition of radiographic structural damage with aTNF agents.17 There is thus an unmet need for new, effective therapies for AS.
Genetic susceptibility and immune dysregulation, with the overexpression of pro-inflammatory cytokines, play important roles in the pathogenesis of AS. Interleukin-6 (IL-6) is a pro-inflammatory cytokine found at elevated levels in AS, and levels correlate with disease activity.18–22 Treatment with the aTNF agent infliximab lowered IL-6 levels in patients with AS, and such reductions correlated with improvements in disease activity and bone mineral density.23 ,24
Tocilizumab (TCZ) is a recombinant, humanised, monoclonal antibody that binds to soluble- and membrane-expressed IL-6 receptors, thereby inhibiting IL-6-mediated signalling.25 The efficacy and safety of TCZ in rheumatoid arthritis (RA) have been demonstrated in randomised-controlled trials.26–30 TCZ is widely approved for the treatment of adult patients with moderately to severely active RA who have had inadequate responses to one or more DMARDs or aTNFs;31 ,32 it is also approved for the treatment of patients with systemic juvenile idiopathic arthritis.31 ,32 TCZ has not been evaluated for AS in a prospective, randomised, clinical trial setting. We conducted two multicentre, double-blind, randomised, placebo-controlled trials—BUILDER-1 and BUILDER-2—designed to investigate the efficacy and safety of TCZ in patients with AS.
Methods
Patient population
Patients eligible for the BUILDER-1 and BUILDER-2 studies were 18 years of age or older; had AS, diagnosed according to the modified New York criteria,33 for ≥3 months; had active disease defined as Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 on a 0–10 cm visual analogue scale (VAS) and spinal pain ≥40 on a 0–100 mm VAS; and had responded inadequately or were intolerant to ≥1 current or previous NSAIDs. Patients in BUILDER-1 were naive to aTNF agents, and elevated C reactive protein (CRP) levels were not required. Patients in BUILDER-2 had responded inadequately to ≥1 aTNF agent administered for ≥3 months and had CRP levels greater than the upper limit of normal (ULN) (0.3 mg/dl).
Study design
BUILDER-1 was a multinational, two-part, parallel-group, phase II–III study. Part 1 (phase II) was conducted at 30 centres in seven countries (Bulgaria, Canada, Czech Republic, Lithuania, Slovakia, South Africa, USA). In part 1, patients were randomly assigned 1:1 to receive TCZ 8 mg/kg or placebo intravenously every 4 weeks for 12 weeks (figure 1). Randomisation was performed centrally and was stratified by geographic region (North America and rest of world). Patient randomisation numbers were provided to an interactive voice or internet response system (IVRS). Patient numbers were allocated sequentially in the order in which patients were enrolled. Medication numbers were assigned by the IVRS at each visit. Participants, treating physicians (or designees), laboratory personnel and sponsor personnel were blinded to study treatment and CRP values during the double-blind periods of the study.
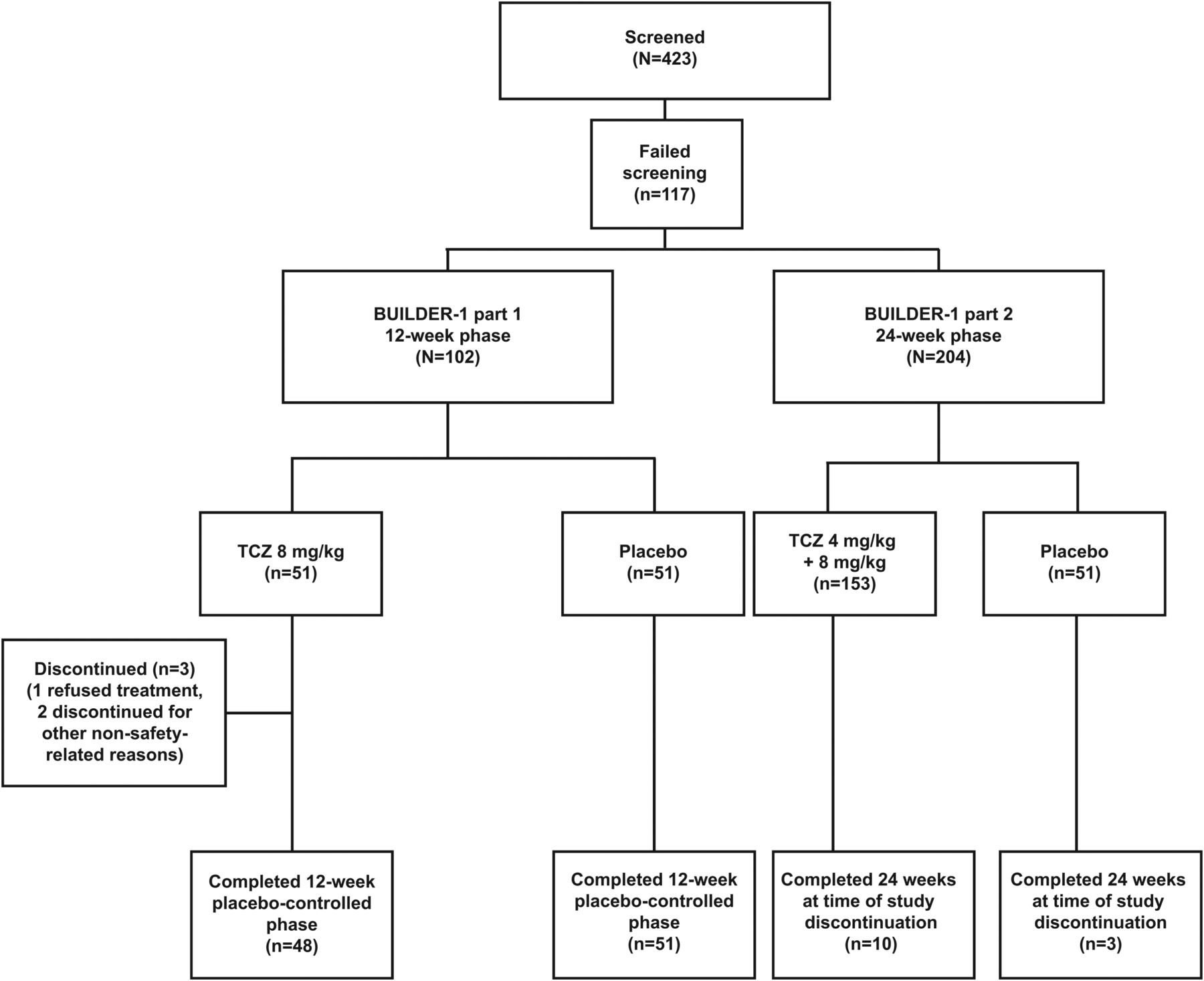
Patient disposition for BUILDER-1 parts 1 and 2 at 12 and 24 weeks. TCZ, tocilizumab.
Part 2 (phase III) was conducted at 59 centres in 12 countries (Australia, Belgium, Bulgaria, Canada, Czech Republic, Lithuania, Poland, Slovakia, South Africa, Spain, UK, USA). Recruitment into part 2 began after enrolment for part 1 was completed. In part 2, patients were randomly assigned 2:1:1 to receive TCZ 8 mg/kg, TCZ 4 mg/kg or placebo intravenously every 4 weeks for 24 weeks (figure 1). Randomisation, in part 2, was stratified by geographic region (North America and rest of world) and by baseline CRP level (≥3× ULN or <3× ULN (3× ULN; 0.9 mg/dl)). Parts 1 and 2 of the study were to be followed by an open-label extension with TCZ 8 mg/kg intravenously every 4 weeks up to an anticipated maximum of 208 weeks.
BUILDER-2 was a phase III study evaluating patients with AS who were aTNF-inadequate responders or who were intolerant of aTNF treatment. Patients were randomly assigned to receive double-blind treatment with TCZ 4 mg/kg, TCZ 8 mg/kg or placebo intravenously every 4 weeks for 24 weeks. In both BUILDER-1 and BUILDER-2, the study protocols were approved by relevant institutional review boards or ethics committees, and written informed consent was obtained from each patient. These studies were carried out in full concordance with the principles of the Declaration of Helsinki and with the laws and regulations of each country in which the research was conducted.
In both studies, NSAIDs, oral corticosteroids (≤10 mg/day prednisone or equivalent) and some DMARDs were permitted. Doses and frequencies of corticosteroids, NSAIDs and DMARDs (methotrexate ≤25 mg/week, sulfasalazine ≤3 g/day, hydroxychloroquine ≤400 mg/day or chloroquine ≤250 mg/day) were to be kept stable during the double-blind period, with modifications allowed only if clinically required for safety reasons.
Efficacy and safety assessments were to be conducted at baseline and at weeks 2, 4, 8, 12, 16, 20 and 24. It was prespecified that a decision regarding continuation or termination of the BUILDER-1 and BUILDER-2 studies was to be made after review of the week 12 efficacy and safety data from part 1 of BUILDER-1.
Efficacy endpoints
The primary efficacy endpoint for the studies was the proportion of patients achieving 20% improvement in the Assessments in SpondyloArthritis international Society (ASAS) Response Criteria (ASAS20).
Secondary efficacy endpoints included the proportion of patients achieving ASAS20 responses at week 24, ASAS40 responses at weeks 12 and 24, ASAS partial remission (values ≤2 in each of the four ASAS parameters: patient global, spinal pain, function, inflammation) at week 12, BASDAI50 responses at weeks 12 and 24 and change in CRP at weeks 2, 12 and 24. Ad hoc exploratory subgroup analyses of the ASAS20 responses in BUILDER-1 part 1 were conducted based on CRP level (<3× ULN, ≥3× ULN), IL-6 level (<40, ≥40 pg/mL), HLA-B27 status (positive, negative), Bath Ankylosing Spondylitis Functional Index (BASFI) score (≤4.5, >4.5–≤6.5, >6.5), presence or absence of peripheral arthritis and Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) score (0, >0). Several secondary and exploratory endpoints in part 2, including MRI and radiographic assessments, were not analysed because of the early termination of the studies.
Safety measures
Safety was assessed in both studies by reporting of adverse events (AEs) and findings on clinical laboratory tests, physical examination, vital sign monitoring, ECGs and chest radiographs.
Sample size and statistical analysis
Sample sizes for BUILDER-1 and BUILDER-2 were determined to provide an adequate safety population for each dose level while ensuring there was adequate power (≥80%) in the analysis of the ASAS20 and ASAS40 responses at week 12. ASAS20 responses of 28% and 59% in the placebo and TCZ arms, respectively, were assumed.
The primary endpoint was analysed using a logistic regression model adjusted for the stratification factor (in part 1) or factors (in part 2) applied at randomisation. Patients who withdrew from the study before the week 12 ASAS20 assessment or for whom week 12 ASAS20 responses could not be determined for any reason were considered ASAS20 non-responders in the primary analysis. Analyses of endpoints and treatment comparisons were performed in a fixed sequential order to control the type 1 error rate for multiple comparisons. The order used was as follows: ASAS20, ASAS40, BASDAI50, ASAS5/6, ASDAS-CRP, ASDAS-ESR, ASAS partial remission, CRP and BASFI.
Results
After a protocol-specified review of the 12-week efficacy and safety data for BUILDER-1 part 1, the study sponsor chose to terminate the BUILDER-1 and BUILDER-2 studies. As a consequence, no meaningful statistical analysis of the efficacy data could be conducted for BUILDER-1 part 1 beyond 12 weeks, BUILDER-1 part 2 or BUILDER-2. Thus, the efficacy results presented here focus on the placebo-controlled, 12-week phase of BUILDER-1 part 1; the statistical analysis is limited to the primary endpoint of ASAS20 response. The safety results include BUILDER-1 part 1 data and pooled data derived from all patients exposed to TCZ in the AS clinical trial programme. The trial was conducted from 21 September 2010 (first patient screened) to 21 December 2011 (last patient visit).
BUILDER-1 part 1 (placebo-controlled phase, 12 weeks): patient population and efficacy
Patient population
A total of 102 patients were enrolled (51 TCZ, 51 placebo); of these, 99 patients from seven countries completed the 12-week placebo-controlled phase (48 TCZ, 51 placebo). Three patients in the TCZ arm discontinued (one refused treatment, two stopped treatment for other non-safety related reasons) (figure 1).
Baseline demographics and disease characteristics
Baseline demographics and disease characteristics were in keeping with the expected study population and were comparable between treatment arms (table 1). Patients were predominantly men and HLA-B27 positive; mean ages were 41.6 and 42.7 years for the TCZ and placebo arms, respectively. Mean disease durations were 5.4 and 7.5 years for the TCZ and placebo arms, respectively. Mean BASDAI scores and mean CRP levels were similar between TCZ and placebo arms.
Baseline demographics and disease characteristics for patients in BUILDER-1 part 1 (placebo-controlled phase, 12 weeks)
Efficacy
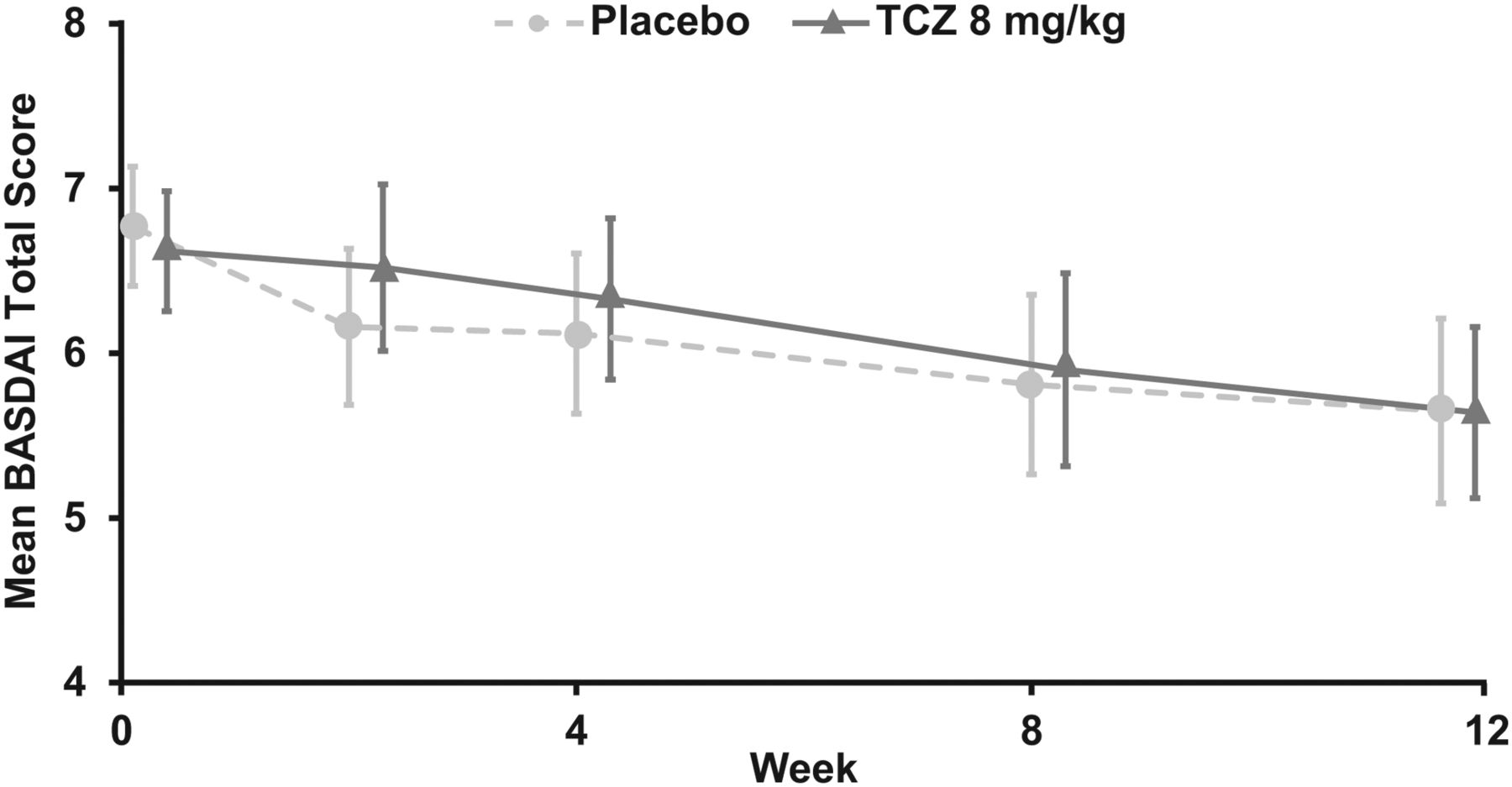
At week 12, the proportion of patients achieving ASAS20 response was similar in the two treatment arms (37.3% and 27.5% in the TCZ and placebo arms, respectively; OR 1.59; 95% CI 0.68 to 3.67; p=0.2823) (figure 2A) as was the proportion of patients achieving ASAS40 response (11.8% and 19.6% in the TCZ and placebo arms, respectively; OR 0.54; 95% CI 0.18 to 1.62; p=0.2694) (figure 2B). During the 12-week, placebo-controlled phase, mean BASDAI scores were similar between the TCZ and placebo arms across all study visits (figure 3). Given that the primary efficacy endpoint was not significantly different no subsequent statistical analysis was conducted.

(A) ASAS20 and (B) ASAS40 response rates at week 12 in BUILDER-1 part 1 (intention-to-treat population; significance was determined using a logistic regression adjusted for region) for the entire group and (C) by CRP levels at baseline. ASAS20, 20% improvement in the Assessments in Ankylosing Spondylitis Response Criteria; CRP, C reactive protein; TCZ, tocilizumab; ULN, upper limit of normal.

BASDAI total score over time in BUILDER-1 part 1 (intention-to-treat population). Error bars represent 95% CIs. Missing components were imputed using last observation carried forward. BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; TCZ, tocilizumab.
Analysis of the change in peripheral arthritis (assessed by the change in swollen joint count, 44 joints (n=31 for TCZ and n=30 for placebo)) and enthesitis (MASES score (n=34 for TCZ and n=38 for placebo)) from baseline to week 12 demonstrated no clinically important difference between the two treatment arms. In patients with ≥1 swollen joint, the median (mean) baseline number of swollen joints was 6.0 (7.0) and 4.0 (7.2) for patients in the TCZ (n=33) and placebo (n=30) arms, respectively. The median (mean) change from baseline to week 12 was −2.0 (−2.2) compared with −1.0 (−2.5) for the TCZ and placebo arms, respectively (see online supplementary figure S1).
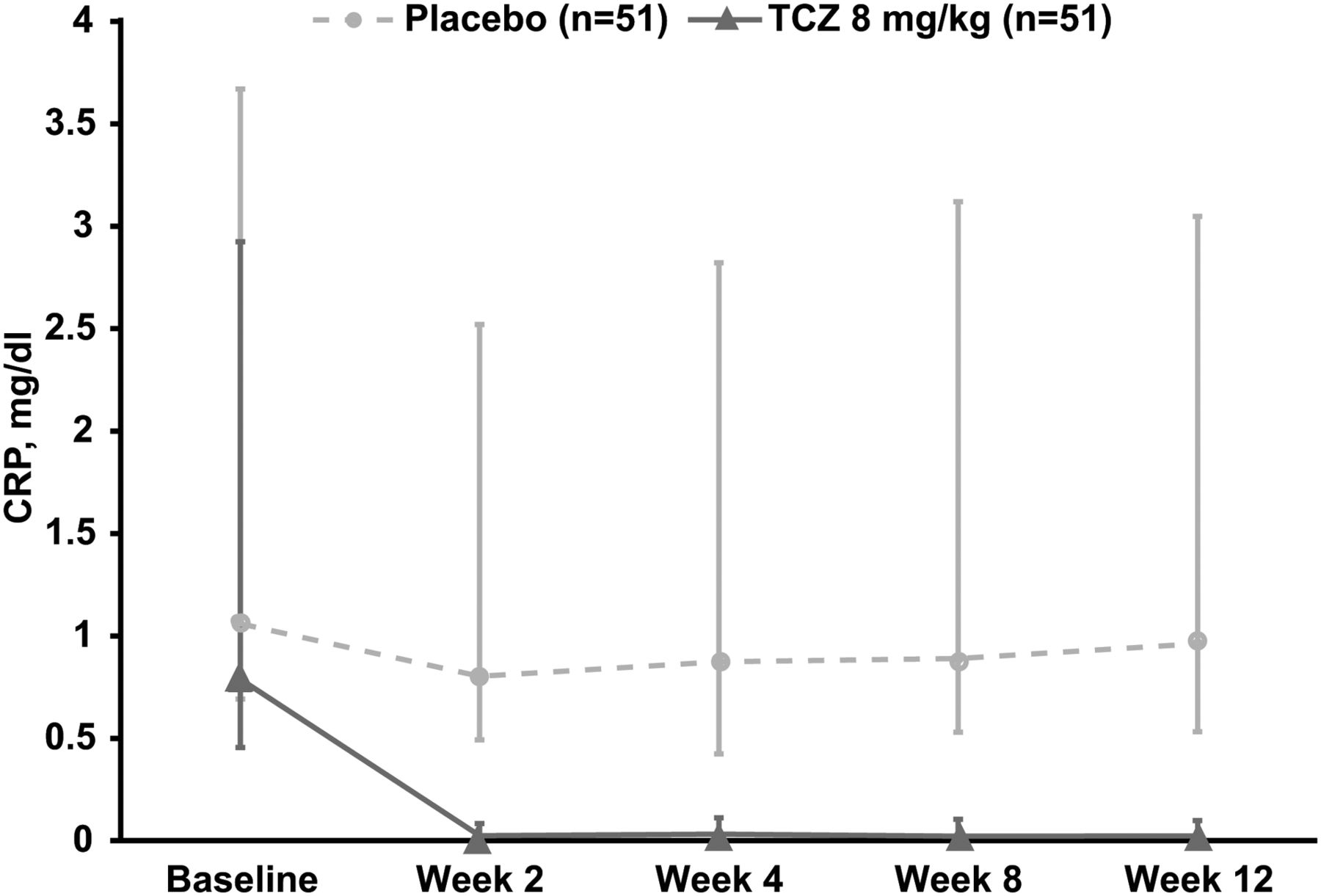
Reductions in mean and median CRP levels from baseline to week 12 were observed in the TCZ arm (−1.34 and −0.73 mg/dl, respectively) but not in the placebo arm (−0.17 and −0.03 mg/dl, respectively) (figure 4). In an exploratory subgroup analysis of patients with high CRP levels (defined as ≥3× ULN) at baseline (n=54), the ASAS20 response rate at week 12 was numerically higher in patients receiving 8 mg/kg TCZ (52.0%) than in those receiving placebo (27.6%). In patients with baseline CRP levels <3× ULN (<0.9 mg/dl) (n=48), ASAS20 response rates were similar in the TCZ (23.1%) and placebo (27.3%) arms (figure 2C). The observed difference in favour of TCZ in patients with higher baseline CRP levels was not observed for ASAS40 response rates (12.0% vs 17.2% for the TCZ and placebo arms, respectively) (figure 2C). Other subgroup analyses using IL-6 level, HLA-B27 status, BASFI score, presence of peripheral arthritis or MASES score did not show a clinically important difference between TCZ and placebo in ASAS20 response rates. Additional exploratory endpoints are reported in online supplementary table S1.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Median change in CRP levels from baseline to week 12 in BUILDER-1 part 1 (intention-to-treat population). Error bars represent IQRs. CRP, C reactive protein; TCZ, tocilizumab.
BUILDER-1 part 2 and BUILDER-2: patient population and efficacy
BUILDER-1 part 2 enrolled 204 patients; all were randomly assigned to treatment, and 203 received treatment (152 TCZ, 51 placebo). BUILDER-2 enrolled 113 patients; all were randomly assigned and received treatment (91 TCZ, 22 placebo). Demographic and baseline characteristics of both patient populations were similar to those reported for BUILDER-1 part 1. Because of the sponsor's decision to terminate the studies, no formal efficacy analyses were performed. Overall, efficacy outcomes appeared consistent with those observed in the placebo-controlled phase of BUILDER-1 part 1, with no differences observed between TCZ and placebo arms for the primary and key secondary endpoints (data not shown). There was no apparent increase in response to TCZ with extended treatment (data not shown).
BUILDER-1 part 1 (placebo-controlled phase, 12 weeks): safety
AEs occurred at similar rates in the TCZ and placebo arms. In all, 30 AEs occurred in 20 of 51 patients in the TCZ arm, and 27 AEs occurred in 13 of 51 patients in the placebo arm (262.4/100 patient-years (PY) (95% CI 177.0 to 374.6) vs 226.9/100 PY (95% CI 149.5 to 330.2), respectively) (table 2). Two serious AEs (SAEs) were reported in two of 51 patients (17.5/100 PY; 95% CI 2.1 to 63.2) in the TCZ arm (cholecystitis and iridocyclitis), and none were reported in the placebo arm. No opportunistic infections, serious infections, AEs leading to study withdrawal or deaths were reported (table 2).
Safety outcomes in BUILDER-1 part 1 (week 12, safety population)
The overall safety profile of TCZ across the entire AS programme was consistent with that observed in the randomised phase of BUILDER-1 (see online supplementary table S2) except for a cluster of anaphylactic and serious hypersensitivity events. Total TCZ exposure in BUILDER-1 and BUILDER-2 (n=355) was 169.9 PY, with overall rates of AEs and SAEs of 207.1/100 PY (95% CI 186.1 to 229.9) and 10.0/100 PY (95% CI 5.8 to 16.0), respectively. Many reported AEs and SAEs were indicative of an exacerbation of AS (see online supplementary table S1).
Four anaphylactic AEs and one serious hypersensitivity AE (allergic reaction during infusion) were reported in BUILDER-1 part 2 and BUILDER-2. All five patients were at three study sites in Bulgaria; no cases occurred outside Bulgaria. All the events occurred within 20–30 min after infusion of TCZ; three patients were receiving 8 mg/kg and two patients were receiving 4 mg/kg. One of the events occurred after the first dose of TCZ; the remaining four events occurred after the second to fifth infusions of drug. The overall rate of anaphylactic AEs in the BUILDER-1 and BUILDER-2 studies was 2.4/100 PY; in Bulgarian sites, this rate was 13.3/100 PY.
Discussion
The use of TCZ for the treatment of patients with AS has been reported in case studies and case series with mixed results.34–39 BUILDER-1 and BUILDER-2 are the first prospective, randomised, placebo-controlled trials designed to assess the efficacy and safety of TCZ in the treatment of AS. The results of BUILDER-1 part 1 (12 weeks) indicated that TCZ was not effective (compared with placebo) in treating the signs and symptoms of AS in NSAID-inadequate responders who were aTNF-naive. This was true in spite of an observed suppression of CRP levels suggesting adequate IL-6R blockade. Although a numerically superior ASAS20 response was observed in the subgroup of patients with a CRP ≥3× ULN (0.9 mg/dl) at baseline who received TCZ, this is difficult to interpret in light of the lack of a parallel response in ASAS40 response rates. In view of the efficacy of TCZ in RA, an effect on peripheral arthritis might have been anticipated, but this was not observed.
The lack of efficacy in the current study was not attributable to any apparent issues with study design, statistical power or study conduct. As such, this negative result highlights that preclinical data may not accurately predict the efficacy of a targeted agent. Other biologics, such as anakinra or abatacept, which have shown efficacy in RA, also failed in AS,40 ,41 suggesting a poorly understood difference in the inflammatory and immune-mediated pathways of different chronic conditions.
Although previous studies have correlated increased IL-6 serum levels with AS disease activity18 ,21 ,42 and reductions in IL-6 during aTNF treatment correlated with improvement in disease activity and spinal inflammation,23 the results of BUILDER-1 part 1 suggest that IL-6 blockade may not be an effective therapeutic approach in AS. A recent study43 of sarilumab, a human monoclonal antibody directed against the α-subunit of the IL-6 receptor complex, also failed to demonstrate efficacy in AS.
Except for the unusually high rate of anaphylactic/serious hypersensitivity AEs, with five serious events occurring at three study sites in Bulgaria, the overall incidences of AEs and SAEs in the current studies were consistent with those observed for TCZ in the RA population.44 This cluster of hypersensitivity cases in Bulgaria was investigated through quality assurance visits at the sites concerned, review of TCZ batches used and review of antidrug antibody data, but we were unable to find an apparent explanation.
Conclusions
TCZ is not an effective treatment for patients with AS. Based on the data observed in our studies and in another placebo-controlled trial, we conclude that despite a pharmacological effect (eg, a relevant decrease in CRP), IL-6 inhibition does not result in a clinically meaningful symptomatic effect on the axial symptoms of spondyloarthritis. Other studies would be required in order to evaluate the symptomatic effect of IL-6 inhibition on other clinical features of spondyloarthritis (eg, peripheral articular involvement, psoriasis, uveitis, inflammatory bowel disease).
Acknowledgments
Our thanks go to all the patients who took part in the BUILDER studies and to all investigators and nursing staff at the participating centres. The authors wish to acknowledge Santo D'Angelo, PhD, MS, and Mark English, PhD, who provided writing services on behalf of F. Hoffmann-La Roche.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tables
Footnotes
Handling editor Tore K Kvien
-
Contributors All authors were involved in the study design, interpretation of the results and the writing of this manuscript. Data collection, data entry and statistical analysis were performed by F. Hoffmann-La Roche. All authors had access to full information from F. Hoffmann-La Roche. All authors agreed on the final content of the submitted manuscript.
-
Funding Funding for the studies was provided by F. Hoffmann-La Roche.
-
Competing interests Joachim Sieper has received grants from Merck Sharp & Dohme, Pfizer, Abbott and Janssen; consulting fees from F. Hoffmann-La Roche, Eli Lilly and Company, Merck Sharp & Dohme, Pfizer, Abbott and Novartis; expert testimony fees from Abbott; and lecture/speakers honoraria from Abbott, Merck Sharp & Dohme, Pfizer and UCB. Benjamin Porter-Brown is an employee of and a stockholder in Roche Products. Liz Thompson is an employee of and a stockholder in Roche Products. Olivier Harari is an employee of and a stockholder in Roche Products. Maxime Dougados has received grants, consulting fees, board membership fees and lecture/speakers honoraria from F. Hoffmann-La Roche, Abbott, Pfizer, UCB and Eli Lilly and Company.
-
Ethics approval In both BUILDER-1 and BUILDER-2, the study protocols were approved by relevant institutional review boards or ethics committees, and written informed consent was obtained from each patient. These studies were carried out in full concordance with the principles of the Declaration of Helsinki and with the laws and regulations of each country in which the research was conducted.
-
Provenance and peer review Not commissioned; externally peer reviewed.