Article Text

Abstract
Objectives To compare over 2 years the safety, efficacy and radiographic outcomes of subcutaneous abatacept versus adalimumab, in combination with methotrexate (MTX), in patients with rheumatoid arthritis (RA).
Methods AMPLE is a phase IIIb, 2-year, randomised, investigator-blinded study with a 1-year primary endpoint. Biologic-naive patients with active RA and an inadequate response to MTX were randomised to 125 mg abatacept weekly or 40 mg adalimumab bi-weekly, both with a stable dose of MTX.
Results Of 646 patients randomised, 79.2% abatacept and 74.7% adalimumab patients completed year 2. At year 2, efficacy outcomes, including radiographic, remained comparable between groups and with year 1 results. The American College Rheumatology 20, 50 and 70 responses at year 2 were 59.7%, 44.7% and 31.1% for abatacept and 60.1%, 46.6% and 29.3% for adalimumab. There were similar rates of adverse events (AEs) and serious adverse events (SAEs). More serious infections occurred with adalimumab (3.8% vs 5.8%) including two cases of tuberculosis with adalimumab. There were fewer discontinuations due to AEs (3.8% vs 9.5%), SAEs (1.6% vs 4.9%) and serious infections (0/12 vs 9/19 patients) in the abatacept group. Injection site reactions (ISRs) occurred less frequently with abatacept (4.1% vs 10.4%).
Conclusions Through 2 years of blinded treatment in this first head-to-head study between biologic disease-modifying antirheumatic drugs in RA patients with an inadequate response to MTX, subcutaneous abatacept and adalimumab were similarly efficacious based on clinical, functional and radiographic outcomes. Overall, AE frequency was similar in both groups but there were less discontinuations due to AEs, SAEs, serious infections and fewer local ISRs with abatacept.
ClinicalTrials.gov Identifier NCT00929864.
- Rheumatoid Arthritis
- DMARDs (Biologic)
- Methotrexate
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
The current recommendations for the management of rheumatoid arthritis (RA) emphasise the early use of methotrexate (MTX) and the addition of biologic disease-modifying antirheumatic drugs (bDMARDs) in patients with an incomplete response to MTX.1–3 The combination of a bDMARD with MTX has demonstrated superior outcomes to either biologic or MTX monotherapy in clinical trials, and is the standard-of-care for patients with active RA.4–6
The currently approved bDMARDs target multiple mechanisms of action (MOAs), including T cell costimulation (abatacept) and tumour necrosis factor-α inhibition (eg, adalimumab), the most widely used agents.7–9 Small molecule DMARDs targeting unique MOAs are also in varying stages of development.10 ,11 In the absence of head-to-head clinical trial data, the question remains how any of these agents with different MOAs compare with respect to clinical efficacy, inhibition of radiographic progression and safety.12 Comparative trials can address this question and are essential to inform evidence-based treatment decisions.9 ,13 ,14 Several recent trials have included two agents in the same study, but these comparisons were either not powered or were made indirectly by comparing both agents to placebo.15–17 Only two trials have included a powered, head-to-head comparison of bDMARDs, Abatacept versus Adalimumab Comparison in Biologic-Naive RA Subjects with Background Methotrexate (AMPLE) and Tocilizumab monotherapy versus adalimumab monotherapy for treatment of rheumatoid arthritis (ADACTA).18 ,19 ADACTA compared tocilizumab monotherapy with adalimumab in patients intolerant or unable to use MTX in a 24-week study, but did not include radiographic outcomes.
AMPLE is a 2-year, phase IIIB, multinational, prospective, randomised study comparing subcutaneous abatacept and adalimumab on stable background MTX in patients naive to bDMARDs and is the only one of these comparative trials to date to also include radiographic assessment.19 Results from the first year of the study revealed comparable onset and magnitude of efficacy, similar inhibition of radiographic damage progression, and generally similar safety.19 The primary endpoint was at 1 year but the blinded study continued for 2 years to provide controlled, comparative assessment of long term safety, efficacy and radiographic outcomes. Here we present the results of the full 2-year AMPLE controlled study period.
Methods
Patients
AMPLE trial design and patient eligibility criteria have been previously described.19 Patients met the 1987 American Rheumatism Association (ARA) criteria for RA, had active disease for ≤5 years despite MTX therapy and were naive to biologic therapy.20
Study design
Patients were equally assigned to receive 125 mg abatacept (Orencia; Bristol-Myers Squibb), administered subcutaneous weekly (without an intravenous loading dose), or 40 mg adalimumab (Humira; Abbott Laboratories), administered subcutaneous every other week, both in combination with a stable dose of MTX (≥15 and ≤25 mg/week or ≥7.5 mg/week if documented intolerance to higher doses). MTX downward titration was allowed at the investigator's discretion only during year 2 which was not to exceed a total decrease of >5 mg/week or go below 7.5 mg/week. No adjustment within 56 days of Day 729 was allowed. Double-blinding of the study drugs was not feasible due to the logistic barrier of masking Humira; patients were not blinded with regard to their study drug. Clinical assessors were blinded to patient treatment and assessed patients’ joints, disease activity and defined adverse event (AE) causality. Study conduct and investigator blinding remained unchanged through the 2-year study period.
Clinical and imaging assessments
Clinical outcomes evaluated included: American College Rheumatology ACR 20, 50, 70 and 90 responses, changes in Disease Activity Score in 28 joints using the C reactive protein level (DAS28-CRP) score, DAS28-CRP <2.6 and ≤3.2, improvement in the Health Assessment Questionnaire–Disability Index (HAQ-DI) ≥0.3 units, and Boolean remission (post hoc analysis).21–23 Efficacy and safety outcomes were evaluated at days 1, 15, 29 and every 4 weeks thereafter during year 1, and every 3 months during year 2.
Plain radiographs of the hands and feet were taken at baseline, year 1 and 2 and scored using the modified Sharp/van der Heijde scoring system.24 Baseline and year 1 radiographs previously reported were reread concurrent with year 2 films by readers blinded to sequence and treatment. Radiographic non-progression was defined as total Sharp (TSS) score ≤ smallest detectable change (SDC); SDC is an estimate of the measurement error between readers of the films.25 Mean changes from baseline in the modified TSS, erosion score (ES) and joint space narrowing score (JSN) were also calculated. Cumulative probability plots were used to assess distribution of radiographic scores.
Safety assessments
Safety assessments were classified using the Medical Dictionary for Regulatory Activities. AEs of special interest included infections, autoimmune events, malignancy and injection site reactions (ISRs). Blood samples for measurement of autoantibodies (antinuclear antibody (ANA) and anti-double stranded DNA (anti-dsDNA)) were collected at baseline, year 1 and year 2.
Statistical analyses
All efficacy analyses were assessed using the intent-to-treat (ITT) populations; all safety analyses were assessed using the as-treated population. The ITT analysis population includes all randomised patients who received at least one dose of medication. The as-treated analysis population includes all randomised patients who received at least one dose of study medication (and patients were grouped on an as-randomised basis unless the patient received incorrect medication for the entire period of treatment). All patients who prematurely discontinued the study after receiving the study drug, regardless of the reason, were considered non-responders at all subsequent visits for ACR and HAQ response analyses. Baseline demographics and clinical characteristics were analysed descriptively for all patients. The primary objective was to demonstrate the non-inferiority of the two treatments as assessed by ACR 20 response at year 1; formal, statistical testing was applied.19
Treatment differences at year 2 were calculated for efficacy assessments with point estimate and 95% CI with no formal statistical testing. Assessments of changes from baseline and construction of CIs for continuous measures were based on analysis of covariance (which included treatment as the main factor, baseline measure and disease activity stratification as covariates). Point estimates and 95% CIs are provided for the difference in adjusted mean change from baseline between the two treatment groups. For mean change in DAS28-CRP scores, HAQ-DI scores and ACR core component scores, missing values were imputed using a last observation carried forward analysis. For patients who discontinued between years 1 and 2, radiographs were obtained at an early termination visit; in these patients, the 2-year data were imputed using linear extrapolation based on assessments performed at baseline and at the time of discontinuation. Subjects without baseline radiographs were excluded from all radiographic analyses.
Post hoc analyses assessed the proportion of patients who achieved ACR/European League Against Rheumatism (EULAR) Boolean-based remission (based on 66 swollen/68 tender joint count).26
Results
Patient disposition and baseline characteristics
A total of 646 patients were randomised and treated: 318 in the abatacept plus MTX group and 328 in the adalimumab plus MTX group (figure 1). The baseline demographics and clinical characteristics have been previously reported and included patients with a mean disease duration of ∼2 years, ∼51 years of age and a mean DAS28-CRP score of ∼5.5 with an equal proportion of patients with DAS28-CRP above and below 5.1 in each group (table 1).19
Baseline demographics and clinical characteristics (intent-to-treat population)*

Disposition of patients over 2 years in the intent-to-treat population randomised to subcutaneous (SC) abatacept or adalimumab, both given in combination with methotrexate (MTX).
Overall, 86.2% (274/318) of the abatacept patients and 82% (269/328) of the adalimumab patients completed year 1 of the study;19 79.2% (252/318) and 74.7% (245/328) completed year 2. The main reasons for discontinuation were AEs (3.5% for abatacept vs 9.1% for adalimumab) and lack of efficacy (6.0% for abatacept vs 4.9% for adalimumab).
Concomitant medications
The dose of MTX (mean±SD) at baseline was 17.5±6.4 mg/week in the subcutaneous abatacept group and 17.3±6.2 mg/week in the adalimumab group; after 2 years of treatment, it was 16.3±4.6 mg/week and 16.3±6.1 mg/week, respectively. Additional concomitant DMARDs (hydroxychloroquine or sulfasalazine) were used at baseline in approximately 15% of both groups; this did not change significantly over the study period. The proportions of patients receiving corticosteroids at any time over 2 years were 65.1% and 64.0%, respectively. More than one course of high-dose corticosteroids was received by 3.5% of the abatacept patients and 3.4% of the adalimumab patients.
Clinical efficacy
ACR responses
The study met the primary objective at year 1 by demonstrating non-inferiority of abatacept to adalimumab by ACR 20 response (64.8% (95% CI 59.5 to 70.0) for abatacept and 63.4% (95% CI 58.2 to 68.6) for adalimumab; estimate of difference 1.8% (95% CI −5.6 to 9.2)).19 ACR 20 responses for both groups were comparable at 4 weeks and remained comparable through 2 years with 59.7% and 60.1% of patients in the abatacept- and adalimumab-treated groups demonstrating an ACR 20 response at year 2. ACR 50, 70 and 90 response rates were also comparable through 2 years (figure 2A and table 2). At 2 years, 30.2% patients in both treatment groups demonstrated a major clinical response (an ACR 70 score maintained for ≥6 months) (table 2).
Clinical outcomes at the end of years 1 and 2 (intent-to-treat population)

Proportions of patients meeting efficacy endpoints in the subcutaneous (SC) abatacept or adalimumab treatment groups over 2 years. (A) Rates of American College Rheumatology (ACR) responses for the intent-to-treat population (N=318 in the SC abatacept-treated group, and N=328 in the adalimumab-treated group). Error bars represent 95% CIs. (B) Disease Activity Score in 28 joints using the C reactive protein level (DAS28-CRP). Data represent mean DAS28-CRP over 2 years. (C) The proportions of SC abatacept- or adalimumab-treated patients who demonstrated the Health Assessment Questionnaire–Disability Index (HAQ-DI) response (improvement of ≥0.3 units from baseline) over 2 years. Error bars represent 95% CIs. Efficacy outcomes through year 1 have been reported earlier.19
Disease activity
At year 2, the DAS28-CRP score (mean±SD) was 3.1±1.5 in the abatacept group and 3.2±1.5 in the adalimumab group (figure 2B); the adjusted mean change from baseline (mean±SE) was –2.4±0.1 and −2.3±0.1, respectively (table 2). A DAS28-CRP score ≤3.2 was achieved by 65.3% (95% CI 59.5% to 71.2%) and 68.0% (95% CI 62.2% to 73.9%) of patients in the abatacept and adalimumab groups, while 50.6% (95% CI 44.4% to 56.8%) versus 53.3% (95% CI 47.0% to 59.5%) achieved a score of <2.6. At year 2, low disease activity using the Clinical Disease Activity Index (CDAI) and the Simplified Disease Activity Index (SDAI) was achieved in 65.6% (95% CI 59.7% to 71.5%) versus 67.6% (95% CI 61.8% to 73.5%) and 65.2% (95% CI 59.3% to 71.1%) versus 69.1% (95% CI 63.3% to 74.9%) of the abatacept- and adalimumab-treated patients, respectively.27 ,28 When remission based on CDAI, SDAI and ACR/EULAR Boolean-based criteria was measured at year 2, the proportions of patients achieving these endpoints were also similar: for CDAI, 32.0% (95% CI 26.2% to 37.8%) versus 30.3% (95% CI 24.6% to 36.1%); for SDAI, 31.2% (95% CI 25.5% to 36.9%) versus 32.5% (95% CI 26.6% to 38.4%); and for ACR/EULAR Boolean-based remission, 20.7% (95% CI 15.7 to 25.7) versus 20.5% (95% CI 15.4 to 25.6) in the abatacept and adalimumab groups, respectively.
Physical function
Improvements in HAQ-DI score were comparable between the two treatment groups. At year 2, the proportion of HAQ-DI responders (defined as an improvement in score ≥0.3) was 54.1% (95% CI 48.6% to 59.6%) and 48.8% (95% CI 43.4% to 54.2%) (figure 2C and table 2). The adjusted mean change to year 2 in the HAQ-DI score (mean±SEM) was −0.60±0.04 and −0.58±0.04 in the abatacept and adalimumab groups, respectively.
ACR core components
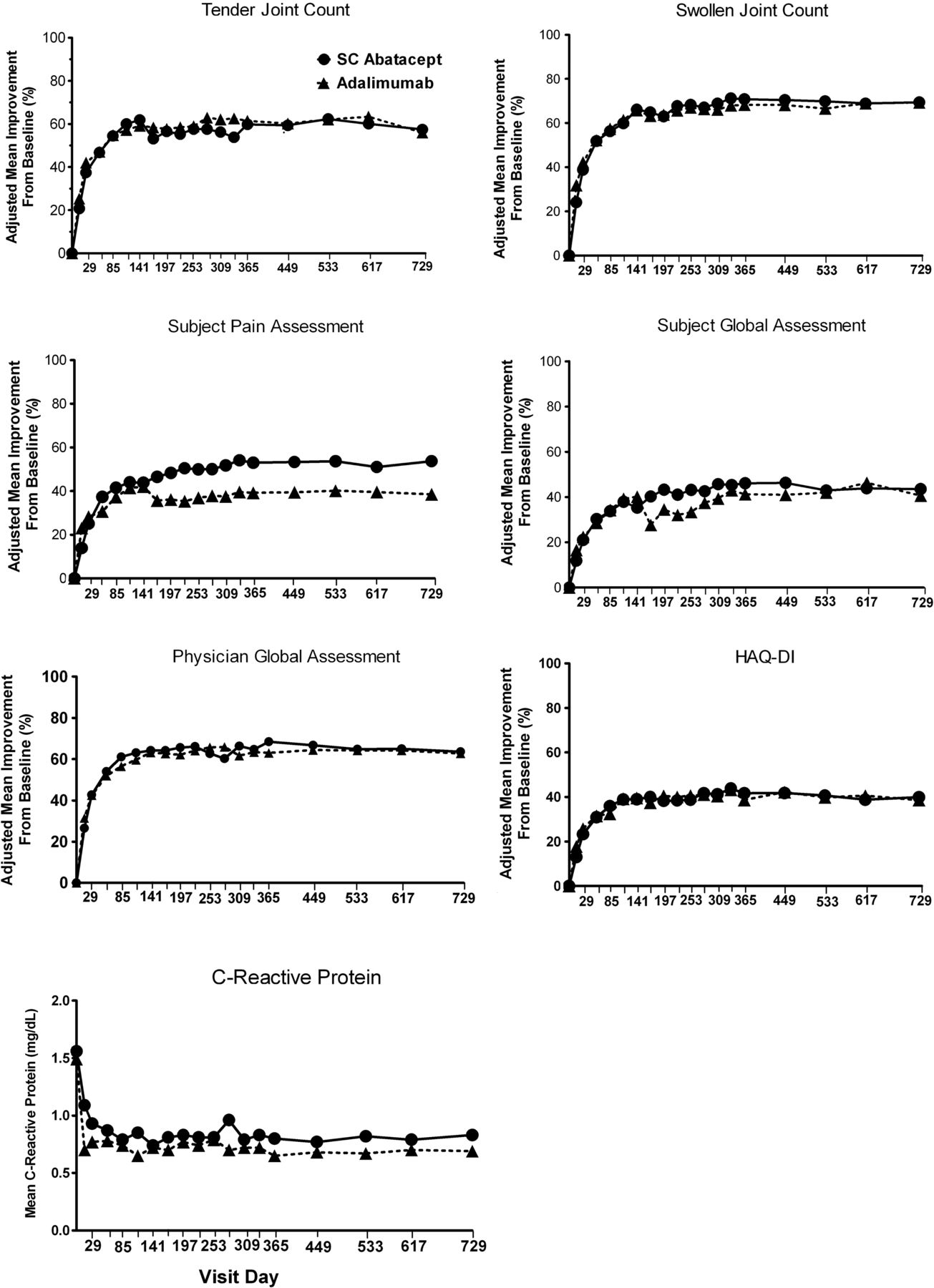
Similar improvements over time were also seen in most ACR core components (figure 3). At year 2, adjusted mean improvements (mean±SEM) for the abatacept and adalimumab groups were: tender joint count: 57.4±6.0% versus 56.0±6.0% (adjusted difference 1.5% (−14.6, 17.5)), swollen joint count: 69.4±2.9% versus 69.3±2.9% (adjusted difference 0.07% (−7.7, 7.8)), HAQ-DI: 39.9±2.9% versus 38.6±3.0% (adjusted difference 1.3 (−6.7, 9.3)), physician's global assessment: 63.6±4.8% versus 62.8±4.7% (adjusted difference 0.9 (−11.9, 13.6)), patient's global assessment of disease activity: 43.5±3.7% versus 40.6±3.6% (adjusted difference 2.9 (−6.9, 12.7)) and patient pain: 53.7±6.2% versus 38.5±6.1% (adjusted difference 15.2 (−1.2, 31.6)). CRP levels (mg/dL, mean±SD) were reduced to 0.80±1.6 versus 0.7±1.3 (adjusted difference 8.3 (–35,7, 52.3)), respectively.

The seven components of the American College of Rheumatology core set of outcome measures were assessed in patients treated with subcutaneous (SC) abatacept or adalimumab over 2 years. Data shown here are adjusted mean change from baseline to 2 years. C reactive protein data shown here are absolute mean values through 2 years. Adjustment based on ANCOVA model with treatment as factor and baseline values, Disease Activity Score in 28 joints using the C reactive protein level (DAS28-CRP) stratification as covariates.
Inhibition of radiographic progression
Paired radiographic images were available at baseline and year 2 for 80.8% (257/318) and 79.3% (260/328) of patients in the abatacept and adalimumab groups. The cumulative probability plot with the distribution of change in total score from baseline to year 2 shows that inhibition of radiographic damage was similar in both treatment groups and included most patients (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Radiographic outcomes in patients treated with subcutaneous (SC) abatacept or adalimumab over 2 years. The cumulative probability plot shows the distribution of change in modified total Sharp/van der Heijde scores of radiographic damage from baseline to 2 years by treatment group.
Through 2 years of treatment, a similar change from baseline in TSS was observed in both groups (mean±SD): 0.9±4.1 versus 1.1±8.7 (table 3). Inhibition of radiographic progression was seen in both component scores (ES 0.4±2.6 vs 0.4±5.0; JSN 0.5±2.2 vs 0.7±3.8) in the abatacept and adalimumab groups, respectively. The changes in mean TSS that occurred during year 2 represented approximately half of that observed during year 1. At year 2, the non-progression rate (change from baseline TSS ≤SDC=2.2) was 84.8% (95% CI 80.4% to 89.2%) versus 83.8% (95% CI 79.4% to 88.3%) in the abatacept and adalimumab groups, respectively. The non-progressor rate using a cut-off of ≤0.5 was 70.8% and 73.1%, respectively.
Radiographic outcomes through 2 years
Safety
At year 2, the cumulative rates of AEs were 92.8% versus 91.5%, and SAEs were 13.8% versus 16.5%, in the subcutaneous abatacept and adalimumab groups (table 4). Discontinuations due to AEs occurred in 3.8% versus 9.5% of the abatacept and adalimumab patients (estimate of difference −5.7 (95% CI −9.5 to −1.9)). Discontinuations due to SAEs occurred in 1.6% versus 4.9% in the abatacept and adalimumab groups (estimate of difference −3.3 (95% CI −9.5 to −1.9)). One death occurred in each treatment group: sudden cardiac arrest in a 66-year-old patient with a history of hypertension in the abatacept group during year 1, and a 75-year-old with a history of hypertension died while hospitalised with acute coronary syndrome in the adalimumab group during year 2.
Safety summary
Infections
Overall, 76.1% of the abatacept patients and 71.3% of the adalimumab patients had an infection over 2 years with nasopharyngitis and upper respiratory tract infections the most frequently reported. During the 2-year study period, serious infections (SIs) occurred in 12 (3.8%) versus 19 (5.8%) patients of which five and 10 occurred during year 2, for abatacept versus adalimumab, respectively (table 4). While none of the SIs in the abatacept group led to treatment discontinuation, nine of the SIs led to adalimumab discontinuation; almost all SIs led to hospitalisation (12 vs 18). Eight opportunistic infections occurred over 2 years, four per group (one case of histoplasmosis (AE) and three cases of oral candidiasis (one SAE, two AE) with abatacept; one case of disseminated histoplasmosis (SAE), two cases of tuberculosis (miliary, pulmonary, both SAEs) and one oral candidiasis (AE) with adalimumab). None of the opportunistic infections in the abatacept group led to discontinuation but both tuberculosis cases in the adalimumab group, which occurred in year 2, led to discontinuation. Herpes zoster was reported in 9 (2.8%) of the abatacept and 6 (1.8%) of the adalimumab patients; none were disseminated or led to discontinuation.
Malignancies
There were no significant differences between the groups in number of malignancies over the 2-year study period. Malignancies occurred in 7 (2.2%) patients in the abatacept group (two squamous cell carcinomas of the skin, one diffuse large B cell lymphoma, one acute myeloid leukaemia, one squamous cell carcinoma of lung, one prostate cancer and one uterine cancer) and 7 (2.1%) patients in the adalimumab group (two basal cell carcinomas, two transitional cell carcinomas, one breast cancer, one malignant melanoma and one small cell lung cancer).
Autoimmune events
Two new autoimmune events occurred in each group during year 2. Over the 2-year study period, autoimmune events were reported in 12 (3.8%) patients in the abatacept group and 6 (1.8%) patients in the adalimumab group (table 4). All of these events were mild or moderate in severity, and three led to discontinuation (two plaque psoriasis in the abatacept and one anti-dsDNA seroconversion in the adalimumab group).
Autoantibody seroconversion
For patients with a baseline negative ANA or anti-dsDNA autoantibody status, ANA seroconversion was observed in 12 (6.3%) versus 24 (14.7%) patients (estimate of difference (95% CI) −8.4 (−15.3 to −1.5)) while anti-dsDNA seroconversion was detected in 0 (0.0%) versus 29 (12.2%) patients (estimate of difference (95% CI) −12.2 (−16.9 to −7.6)) in the abatacept and adalimumab groups, respectively. No cases of lupus-like syndrome were reported.
Local injection site-related events
The proportion of patients with local ISRs during year 1 was a prespecified secondary endpoint for the study; they occurred in significantly fewer patients in the abatacept than the adalimumab group (3.8% vs 9.1%; p=0.006).19 In the cumulative 2-year study period, ISRs were reported in 13 versus 34 patients (4.1% vs 10.4%; difference from adalimumab: −6.3 (95% CI (−10.2 to −2.3); table 4). Differences in intensity were also reported, with severity categorised as 12 mild/one moderate in the abatacept and 27 mild/six moderate/one severe in the adalimumab group. No patients in the abatacept group discontinued due to ISRs, while three patients discontinued in the adalimumab group, all during year 1.19
Discussion
Through 2 years of treatment in this first, head-to-head trial in patients with active RA on background MTX, most subjects who entered the study tolerated the agents well and clinically benefited. Overall, subcutaneous abatacept demonstrated similar clinical efficacy and inhibition of radiographic progression to adalimumab. While the frequency of AE was similar in both groups, there were fewer discontinuations due to AEs, SAEs and SIs and fewer local ISRs in patients treated with abatacept.
A high proportion of patients completed the 2-year study. While the primary endpoint was at 1 year, study conduct remained unchanged with investigator blinding maintained through 2 years. We previously reported the similar onset and kinetics of response through 1 year.19 The 2-year data demonstrate sustained, comparable efficacy across all clinical measures including remission and physical function; these results are consistent with those seen in other studies of abatacept and adalimumab.4 ,16 ,29–34
Radiographic assessment is an important outcome in RA trials; progression of joint damage correlates over the long term with disease activity and disability.25 ,35 ,36 In AMPLE, approximately 80% of the patients had paired films available for radiographic assessment providing a robust data set for long term assessment. Results from year 2 demonstrate continued inhibition of radiographic progression in both treatment groups when compared with year 1. These findings are consistent with previous abatacept and adalimumab studies.29 ,31 Combined with the overlapping cumulative probability curves and comparable radiographic non-progression rates, this shows that through 2 years both agents were similarly effective at inhibiting radiographic progression.
Although the rates of AEs were similar, there were fewer SAEs and fewer discontinuations due to AEs or SAEs in the abatacept group, and in both cases the CIs for the estimates of difference did not cross zero. There were also fewer SIs that led to discontinuation or hospitalisation with abatacept. There were two cases of tuberculosis during year 2 in the adalimumab arm, both of which led to patient discontinuation. Malignancies occurred with similar frequency in both groups. There were more autoimmune AEs observed in the abatacept arm; none of these were considered serious by the investigator. There were less ISRs in the abatacept group up to year 2. Overall, the rates and types of safety events observed were consistent with those reported previously in other, independent clinical trials.16 ,37–40
This study reflects the advantages and limitations of head-to-head clinical trial design. Unlike indirect or statistics-based comparisons of RA patients which can have limited utility because of differences in patient populations and other factors, AMPLE was designed to enrol and directly compare subjects head-to-head in the same study.41–43 For comparative trials to be of value, they must enrol subjects and use therapies that are similar to those used in routine clinical practice.44–46 Adalimumab, combined with the standard-of-care, MTX, is one of the most common therapeutic choices for patients with RA with an incomplete response to MTX; therefore, it is an appropriate comparator.
AMPLE used a single-blinded, rather than double-blinded, design because of an inability to mask commercially acquired Humira; it was also impractical to require weekly visits for blinded injections over 2 years. However, the study did use blinded clinical assessors and radiographic readers to mitigate this limitation, and the similar patient- and physician-reported outcomes support this approach.
To date, AMPLE is the largest head-to-head RA trial comparing two bDMARDs, the first to include radiographic outcomes and the first with blinded, controlled data out to 2 years. The 2-year data further support the comparable efficacy of abatacept and adalimumab, including inhibition of radiographic progression and physical function, previously reported at 1 year. In addition, the data shown here demonstrate further differences in safety through year 2 including differences in the rates of SIs, SAEs and discontinuations due to AEs and SAEs. This study demonstrates that over 2 years of blinded treatment, abatacept and adalimumab achieved similar clinical efficacy and radiographic inhibition, with some notable differences in safety. As such, based on efficacy alone, these two agents should be considered similar for the treatment of RA patients with an inadequate response to MTX. Furthermore, this study demonstrates the value of head-to-head trials in RA and provides data for physicians to make evidence-based treatment choices.
Acknowledgments
We thank the patients and all the investigators who participated in the study. Professional medical writing and editorial assistance were provided by Anu Santhanagopal, PhD, of Bristol-Myers Squibb.
References
Footnotes
-
Handling editor Tore K Kvien
-
Contributors All authors meet the following conditions: (1) conception and design, acquisition of data OR analysis and interpretation of data, (2) drafting the article OR revising it critically for important intellectual content, and (3) final approval of version submitted for publication.
-
Competing interests MS is a consultant and speaker for Bristol-Myers Squibb and Abbott. MW has received grant/research support and is a consultant for Bristol-Myers Squibb and Abbott, and provides expert testimony to Abbott. RV has received grant support from UCB, Pfizer, Novartis, Eli Lilly, Takeda, Bristol-Myers Squibb and Centacor. DvH is a consultant for Abbott, Bristol-Myers Squibb, Amgen, AstraZeneca, Centocor, Chugai, Eli-Lilly, GSK, Merck, Novartis, Otsuka, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, UCB and Wyeth, and is director of Imaging Rheumatology bv. GC is a consultant for Bristol-Myers Squibb, Pfizer, Abbott, and received grant support from Pfizer. AE and MM are employees of Bristol-Myers Squibb and hold stock at BMS. RF is a consultant for Bristol-Myers Squibb, Abbott, Amgen, Pfizer, UCB, Jansen, Roche, Eli-Lilly, Sanofi-Aventis, Novartis, Lexicon, Vertex and GSK, and has received grant/research support from Bristol-Myers Squibb and Abbott.
-
Funding Bristol-Myers Squibb, Princeton, New Jersey, USA, funded the AMPLE trial and provided editorial and submission assistance for this manuscript.
-
Ethics approval The study was conducted in accordance with the Declaration of Helsinki and was consistent with International Conference on Harmonisation Good Clinical Practice. The protocol and patient's informed consent received institutional review board/independent ethics committee approval prior to initiation of the study.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Data reported in this manuscript will be shared if needed.