Article Text

Abstract
Objective To investigate whether biologic-free remission can be achieved in patients with early, active axial spondyloarthritis (SpA) who were in partial remission after 28 weeks of infliximab (IFX)+naproxen (NPX) or placebo (PBO)+NPX treatment and whether treatment with NPX was superior to no treatment to maintain disease control.
Method Infliximab as First-Line Therapy in Patients with Early Active Axial Spondyloarthritis Trial (INFAST) Part 1 was a double-blind, randomised, controlled trial in biologic-naïve patients with early, active, moderate-to-severe axial SpA treated with either IFX 5 mg/kg+NPX 1000 mg/d or PBO+NPX 1000 mg/d for 28 weeks. Patients achieving Assessment of SpondyloArthritis international Society (ASAS) partial remission at week 28 continued to Part 2 and were randomised (1:1) to NPX or no treatment until week 52. Treatment group differences in ASAS partial remission and other efficacy variables were assessed through week 52 with Fisher exact tests.
Results At week 52, similar percentages of patients in the NPX group (47.5%, 19/40) and the no-treatment group (40.0%, 16/40) maintained partial remission, p=0.65. Median duration of partial remission was 23 weeks in the NPX group and 12.6 weeks in the no-treatment group (p=0.38). Mean Bath Ankylosing Spondylitis Disease Activity Index scores were low at week 28, the start of follow-up treatment (NPX, 0.7; no treatment, 0.6), and remained low at week 52 (NPX, 1.2; no treatment, 1.7).
Conclusions In axial SpA patients who reached partial remission after treatment with either IFX+NPX or NPX alone, disease activity remained low, and about half of patients remained in remission during 6 months in which NPX was continued or all treatments were stopped.
- Spondyloarthritis
- NSAIDs
- TNF-alpha
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are first-line treatments for patients with axial spondyloarthritis (SpA) or ankylosing spondylitis (AS). For patients who fail NSAID treatment or cannot tolerate it, tumour necrosis factor (TNF) α blockers are recommended1 because they reduce inflammation, control disease activity and improve patient functioning and quality of life.2–5 In the Infliximab as First-Line Therapy in Patients with Early Active Axial Spondyloarthritis Trial (INFAST) study,6 patients with axial SpA who had not failed NSAIDs or were NSAID-naïve showed substantial benefit from combined treatment with NSAIDs and the TNF-antagonist infliximab (IFX). At month 6, partial remission was achieved in 62% and 35% of patients treated with IFX+naproxen (NPX) or placebo (PBO)+NPX, respectively.
For patients with axial SpA who have achieved remission, the best strategy for maintenance of remission is not known. In patients with rheumatoid arthritis (RA), several studies have shown that biologic-free remission, either with or without continuous treatment with methotrexate, can be achieved if patients are treated early enough in the course of the disease.7–9 In patients with AS, studies have found that response to TNF antagonists may be sustained over several years, but that response is lost soon after cessation of treatment.10 ,11 Few studies have evaluated the long-term effects of TNF-antagonist treatment in patients with early axial SpA, and it is unclear how long biologic-free remission is likely to be maintained or whether NSAID treatment might aid in maintenance of remission after anti-TNF treatment is discontinued.
The primary goals of INFAST Part 2, the 6-month follow-up study reported here, were to explore whether biologic-free remission can be achieved in patients with early axial SpA and to determine whether continued treatment with NPX was superior to discontinuing all treatments in order to maintain disease control for 6 months.
Methods
Study design and patients
Part 1 of INFAST (Protocol P05336, NCT00844805) was a multicentre, double-blind, randomised, controlled trial of IFX+NPX vs PBO+NPX treatment in patients aged 18–48 years with early, moderate-to-severe axial SpA (see online supplementary figure S1). The design of Part 1 of the INFAST study has been reported elsewhere.6 Briefly, patients enrolled in INFAST Part 1 had been diagnosed with axial SpA of less than 3 years’ duration, were naïve to biologics and were either NSAID-naïve at baseline or had been treated with not more than two-thirds of the maximal recommended dose during the 2 weeks prior to the screening visit. Patients were randomised (2:1) to receive 28 weeks of treatment with either intravenous (IV) IFX 5 mg/kg (weeks 0, 2, 6, 12, 18 and 24)+NPX 1000 mg/d or IV PBO+NPX 1000 mg/d.
In Part 2, the follow-up period of INFAST that is the focus of this report, patients who had achieved Assessment of SpondyloArthritis international Society (ASAS) partial remission at week 28 (defined as reaching ≤20 mm on a scale of 100 mm in all four ASAS domains) were eligible to continue to the follow-up phase. IFX treatment was stopped, and patients were randomised to receive either NPX or no treatment (1:1 ratio) until week 52. For the NPX group, open-label NPX was administered at the dose each patient had received prior to week 28 (1000 mg/d or 500 mg/d if the higher dose was not tolerated). A computer-generated randomisation list was created by the sponsor and held by the central randomisation centre, which was contacted by the site to assign treatment to each patient as they enrolled. Randomisation in Part 2 was stratified by randomised treatment assignment in INFAST Part 1. Data were collected from October 2009 to September 2011.
Patients with flares, defined as Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥3 cm on a 10-cm visual analogue scale during two consecutive visits within 1–3 weeks of each other between weeks 28 and 52, had a final MRI and were discontinued from the study.
Outcome measures
The primary outcome of INFAST Part 2 was the percentage of patients who maintained ASAS partial remission until week 52. Secondary outcomes included the percentage of patients with disease flare (as defined above) and several other measures of disease activity, inflammation, clinical symptoms, functioning and quality of life (Ankylosing Spondylitis Disease Activity Score (ASDAS), BASDAI, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), Bath Ankylosing Spondylitis Functional Index (BASFI), Bath Ankylosing Spondylitis Metrology Index (BASMI) and EuroQoL 5D Health Questionnaire (EQ-5D)) were assessed at several points throughout the study.
Adverse events and other safety measures were also collected.
Statistical analysis
No sample size calculation was done for this exploratory follow-up study because the sample was determined by the number of patients in remission at the end of Part 1 of INFAST. Anticipating that approximately 60 patients would be in remission at the end of Part 1, with 30 patients per group, the study had an estimated 87% power to detect a 42% absolute difference between the treatment groups in the percentage of patients with ASAS partial remission at week 52 using a Fisher exact test with α set at 0.05. This was considered acceptable because a small effect of NPX would probably not justify continued use of NPX to maintain remission after stopping a biologic treatment.
The intent-to-treat (ITT) population for INFAST Part 2 included all subjects who were randomised in the follow-up phase and had at least one efficacy assessment after week 28. The safety population included all patients who were randomised in the follow-up phase. Adverse events were analysed descriptively.
Treatment group differences in categorical efficacy measures, including the primary efficacy outcome, were analysed using Fisher exact tests. α was set at 0.05. Patients who discontinued early were considered to be non-responders in the analysis of ASAS partial remission. No other data were imputed. Continuous measures were analysed using analysis of covariance with baseline values as covariates. Duration of ASAS partial remission (in weeks) was assessed using the Kaplan–Meier method with a log-rank test of the difference between treatment groups. Predictors of duration of remission were explored using Cox regression models.
Results
Patient disposition
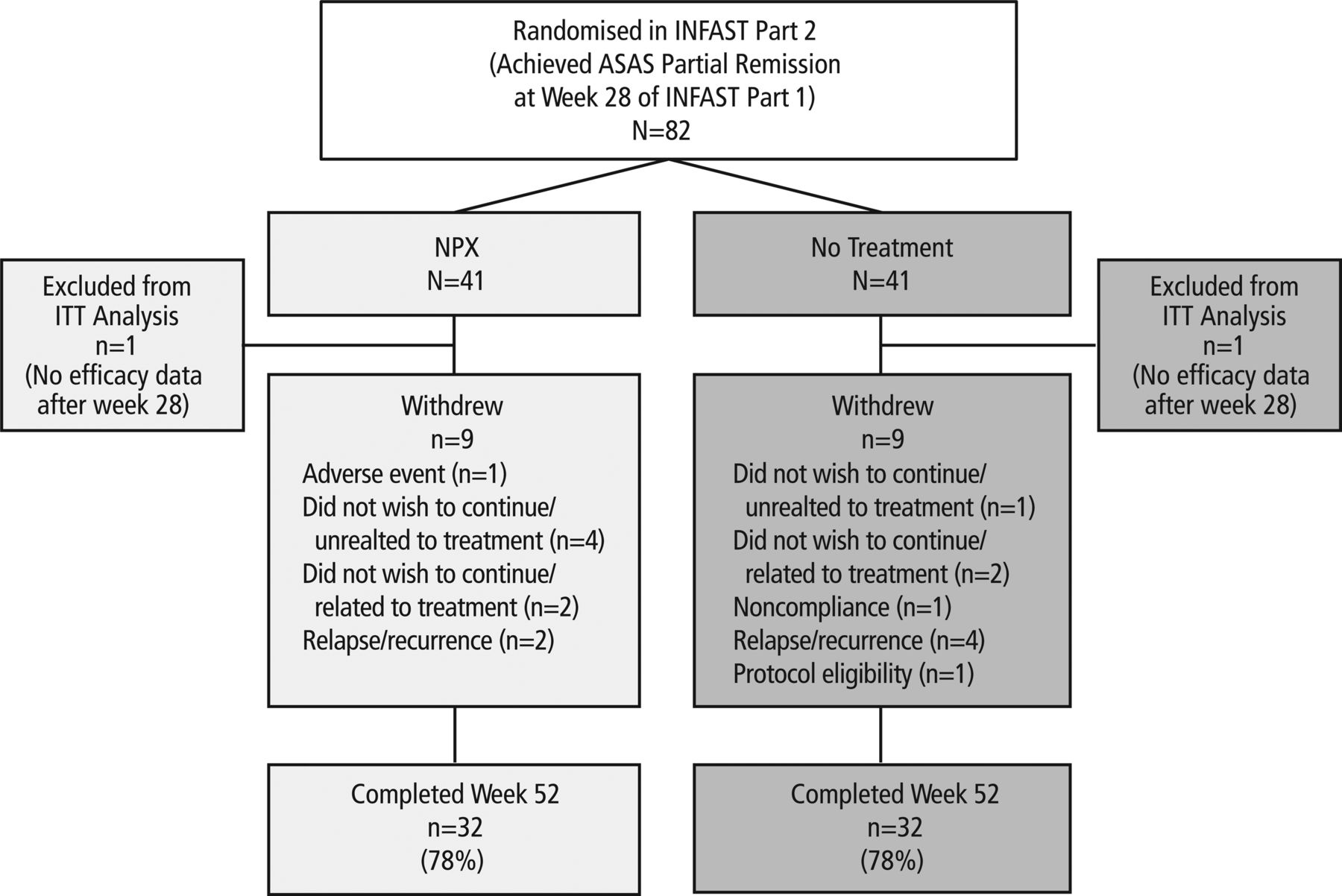
In Part 1 of INFAST, 158 patients were randomised, and 141 completed the study to week 28. In Part 2, the follow-up phase that included only patients who achieved ASAS partial remission at week 28, 41 patients were randomised to NPX and 41 to no treatment (figure 1). The ITT population included 40 patients in each treatment group; 1 patient was excluded from each treatment group because they had no efficacy data after week 28. Each treatment group in Part 2 had 31 patients from the IFX+NPX arm and 9 patients from the PBO+NPX arm in Part 1; the greater number of patients from the IFX+NPX arm reflected the 2:1 randomisation ratio to IFX+NPX vs PBO+NPX in Part 1, and also the greater number of patients who reached partial remission in the IFX+NPX arm in Part 1. Most patients (32/41, 78%) in each of the treatment arms in Part 2 completed week 52 of the trial.

Patient disposition for INFAST Part 2. ASAS, Assessment of SpondyloArthritis international Society; INFAST, Infliximab as First-Line Therapy in Patients with Early Active Axial Spondyloarthritis Trial; ITT, intent-to-treat; NPX, naproxen.
Baseline characteristics
For patients who entered INFAST Part 2, the treatment groups were similar in demographic and clinical characteristics at baseline of INFAST Part 1 (week 0) and clinical characteristics at baseline of INFAST Part 2 (week 28) (table 1). Patients were selected for INFAST Part 2 only if they had reached partial remission in INFAST Part 1, and therefore, disease activity at the beginning of the follow-up phase was low. For example, mean BASDAI scores were 0.7 and 0.6 in the NPX and no-treatment groups, respectively, at week 28, an improvement from a mean of approximately 6.0 at baseline of INFAST Part 1 (week 0).
Demographic and baseline characteristics for patients in INFAST Part 2 (all randomised patients)
Exposure and adherence
In the NPX group, the average daily dose of NPX was 936.6 mg, with 99.4% adherence to the number of doses, regardless of dose amount.
Efficacy
At week 52, similar percentages of patients in the NPX group (47.5%, 19/40) and the no-treatment group (40.0%, 16/40) met the ASAS partial remission criteria, p=0.65 (figure 2A). Overall, the percentage of patients in partial remission decreased steadily during the 6-month follow-up period. At each assessment point, similar percentages of patients in the NPX group and the no-treatment group maintained ASAS partial remission (p>0.05 at each time point). The median duration of partial remission was 23 weeks (95% CI 14.43 to 25.14) in the NPX treatment arm and 12.6 weeks (95% CI 10.71, upper bound not estimable) in the no-treatment group, a difference that was not statistically significant (p=0.38).

Percentage of patients in INFAST Part 2 who had ASAS partial remission (A) and low disease activity (BASDAI<3 in patients with nonmissing data) (B) at each visit. ASAS, Assessment of SpondyloArthritis international Society; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; ET, early termination; INFAST, Infliximab as First-Line Therapy in Patients with Early Active Axial Spondyloarthritis Trial; NPX, naproxen; Wk, week.
Although the percentage of patients in partial remission decreased steadily over the 6-month follow-up period, few patients in either treatment group met the criteria for disease flare (NPX, 1/40, 2.5% and no treatment, 3/40, 7.5%; p=0.62) and the vast majority of patients (83% to 94%) in both groups maintained a state of low disease activity (BASDAI<3) at all visits (figure 2B).
All efficacy measures indicated some worsening from week 28 to week 52, but overall disease activity, as measured by BASDAI and ASDAS, remained very low in both the NPX and no-treatment groups (table 2). BASMI, BASFI, ESR, CRP and EQ-5D showed similar patterns (table 2), with no statistically significant differences between the treatment groups.
Efficacy outcomes by treatment group
Figure 3 illustrates the gains that were made in the first 28 weeks and maintained over the 6-month follow-up period by showing the data over the full-year study period for the group of patients who participated in Part 2. For example, BASDAI scores decreased substantially for patients at the start of treatment in INFAST Part 1. In Part 2, after patients were randomised to NPX or no treatment, the NPX group appeared to have a slightly lower BASDAI at each visit. Patterns were similar for ASDAS based on CRP (ASDAS-C), CRP and BASFI.

Mean BASDAI (A), ASDAS-C (B), CRP (C) and BASFI (D) from baseline to week 52 for patients who participated in INFAST Part 2. aDuring the follow-up period, patients with ASAS partial remission were assigned to either NPX or no treatment, with assignments stratified by initial treatment group. ASAS, Assessment of SpondyloArthritis international Society; ASDAS-C, Ankylosing Spondylitis Disease Activity Score based on CRP; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; CRP, C-reactive protein; IFX, infliximab; NPX, naproxen; PBO, placebo; VAS, visual analogue scale; Wk, week.
Figure 4 shows BASDAI and ASDAS-C scores for the Part 2 participants, grouped according to their randomised assignments in both Part 1 and Part 2. Patients receiving IFX+NPX in Part 1 showed greater improvement than patients receiving PBO through week 34. By week 52, differences were no longer evident between the groups. Although it is useful to see the overall patterns across groups in Part 2, it should be noted that the numbers of subjects in these four groups are small.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
BASDAI (A) and ASDAS-C (B) by treatment sequence and visit for patients who participated in Part 2. aDuring the follow-up period, patients with ASAS partial remission were assigned to either NPX or no treatment, with assignments stratified by initial treatment group. ASAS, Assessment of SpondyloArthritis international Society; ASDAS, Ankylosing Spondylitis Disease Activity Score; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; IFX, infliximab; NPX, naproxen; PBO, placebo; Wk, week.
Single and two-factor logistic regression analyses examined predictors of sustained remission at week 52. Regardless of treatment in Part 1 of INFAST, disease duration in years was the only characteristic that had a robust association with remission at week 52. In the single-variable analysis, the estimate was −1.673 (SE=0.572, p=0.03), indicating that patients with shorter disease duration were more likely to have sustained remission than patients with longer disease duration.
Safety
During the follow-up period, the percentage of patients with treatment-emergent adverse events (TEAEs) was higher in the no-treatment arm (41.5%; 17/41) than in the NPX arm (26.8%; 11/41). The most frequently reported TEAEs in the NPX group were rhinitis and intervertebral disc protrusion (each in two patients). The most frequently reported TEAEs in the no-treatment group were nasopharyngitis (three patients), bronchitis (two patients) and respiratory tract infection (two patients). During the follow-up period, one serious TEAE (carcinoma in situ) was reported in the no-treatment group (the patient had received IFX+NPX in INFAST Part I), and one TEAE leading to early withdrawal (fatigue) occurred in the NPX group. No deaths occurred.
Discussion
In this study of patients with early, active, moderate-to-severe axial SpA who had reached ASAS partial remission following 28 weeks of treatment with IFX+NPX or PBO+NPX, remission was maintained at week 52 by similar percentages of patients who either stayed on NPX therapy (47.5%) or in whom all treatments were stopped between weeks 28 and 52 (40.0%). Although the rate of partial remission, a stringent criterion for disease activity, decreased steadily over the follow-up period, other measures of disease activity indicated that most of the substantial improvement achieved over the first 28 weeks of treatment was well maintained during the follow-up period. For example, mean BASDAI across treatment groups was approximately 6 at baseline of INFAST Part 1, approximately 0.6 at baseline of INFAST Part 2 (week 28) and approximately 1.5 at week 52. Approximately 90% of patients had BASDAI<3 at week 52. As assessed by several other measures of disease activity, inflammation and functioning (eg, ASDAS-C, CRP and BASFI) improvements achieved during the initial treatment phase of INFAST were generally maintained during the 6-month follow-up period, and very few flares were observed.
One issue of importance to patients, clinicians and payers is whether patients with axial SpA who are treated with TNF-α antagonists can stop therapy without experiencing disease flare. Although studies in AS populations have shown that patients continue to respond to TNF-antagonist treatment over periods of several years, available evidence indicates that nearly all patients will flare once treatment is stopped,10 ,11 suggesting that the biologic-free remission rates found in INFAST would be unlikely in patients with established AS and longer disease duration. In the current study's population of patients with early disease, 78% of the patients in the follow-up study had received IFX+NPX in the initial treatment phase of INFAST. When all IFX treatment was stopped at week 28, almost half of the patients maintained partial remission to week 52. Overall, the data suggest that both combination treatments with IFX+NPX and NPX monotherapy in the first 6 months had long-lasting benefits in those patients who reached partial remission, with few patients in any treatment group experiencing disease flares.
The initial positive responses to treatment in INFAST Part 1 and the well-maintained response shown in INFAST Part 2 are among the highest rates of response for studies of TNF-α antagonist treatment in patients with axial SpA.5 ,12–14 The high response rates are possibly due to the very short disease duration of the population, patient selection for signs of inflammation on MRI and the intensified treatment during Part 1 (either a full dose of NPX or NPX+IFX) in a population of patients that had not yet failed NSAIDs or were naïve to NSAIDs at study entry. Patients in INFAST reported a mean of fewer than 2 years since the onset of symptoms, and previous studies4 ,12 ,15 have shown that short disease duration is a predictor of better response to TNF-α antagonists in patients with axial SpA and AS. Shorter disease duration was associated with sustained remission in our study, which may indicate the existence of a window of opportunity for treatment of axial SpA early in the course of disease, as has been described in early RA.16 This is the first trial in axial SpA demonstrating that a remission-induction strategy in patients with very early disease can actually keep these patients in a state of low disease activity, even without medication. One limitation of this study is that it is not known whether these results could be achieved after stopping medication in a population of patients who had already failed NSAIDs.
Although there was a tendency for slightly numerically better outcomes in patients who received NPX rather than no treatment during the follow-up period, none of these differences were statistically significant. It is difficult to draw any conclusions about the possible superiority of NPX because the follow-up phase of INFAST was an exploratory study that was underpowered to detect smaller differences between the NPX and no-treatment groups. Although significant clinically relevant differences could have been missed because of the small sample size, the small differences between the two groups detected here are likely to be too small to be of clinical significance even if a statistically significant difference had been shown in a study with greater power. The open-label nature of the study might be considered a weakness that could bias the results in favour of the NPX group, but the NPX group was not superior to the no-treatment group, suggesting that the open-label design should not influence the overall interpretation of the data.
The results of the INFAST follow-up study demonstrated that patients with early, active, moderate-to-severe axial SpA who achieved partial remission during 28 weeks of IFX+NPX or PBO+NPX treatment had similar rates of partial remission whether they received NPX or no treatment during 6-months of follow-up. Overall, patients had low disease activity and very few flares during the follow-up period, whether they received NPX or no treatment at all. Whether such a low level of disease activity could be maintained beyond 6 months is an important question for future studies.
Acknowledgments
Medical writing assistance was provided by Ellen Stoltzfus, PhD, and Kathy Claussen, PhD, of JK Associates, Inc., Conshohocken, PA. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure
Footnotes
Handling editor Tore K Kvien
-
Contributors All authors are responsible for the work described in this paper and were involved in at least one of the following: conception, design, acquisition, analysis, statistical analysis and interpretation of data. All authors were involved in drafting the manuscript and/or reviewing the manuscript for important intellectual content. All authors provided final approval of the final version.
-
Funding Financial support for this study was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ.
-
Competing interests JS: Grant/Research support from Abbott, Merck, Pfizer, Janssen; Consultant for Abbott, Merck, Pfizer, UCB, Roche, Lilly, Novartis; Speakers Bureau for Merck, Abbott, Pfizer, UCB. JL: Honoraria fees from Abbott, BMS, MSD, Pfizer, Roche, Astra Zeneca. JW: None declared. MR: Consultant and Speakers Bureau for Abbott, BMS, MSD, Pfizer, UCB; Speakers Bureau for Roche. VM: None declared. LM: None declared. S-HP: None declared. Y-WS: None declared. RY: Employee of Merck. DC: Employee of Merck. NV: Employee of Merck.
-
Ethics approval Local review boards.
-
Provenance and peer review Not commissioned; externally peer reviewed.