Article Text

Abstract
Objectives Mavrilimumab, a human monoclonal antibody targeting the alpha subunit of the granulocyte-macrophage colony-stimulating factor receptor, was evaluated in a phase 2 randomised, double-blind, placebo-controlled study to investigate efficacy and safety in subjects with rheumatoid arthritis (RA).
Methods Subcutaneous mavrilimumab (10 mg, 30 mg, 50 mg, or 100 mg) or placebo was administered every other week for 12 weeks in subjects on stable background methotrexate therapy. The primary endpoint was the proportion of subjects achieving a ≥1.2 decrease from baseline in Disease Activity Score (DAS28-CRP) at week 12.
Results 55.7% of mavrilimumab-treated subjects met the primary endpoint versus 34.7% placebo (p=0.003) at week 12; for the 10 mg, 30 mg, 50 mg, and 100 mg groups, responses were 41.0% (p=0.543), 61.0% (p=0.011), 53.8% (p=0.071), and 66.7% (p=0.001) respectively. Response rate differences from placebo were observed at week 2 and increased throughout the treatment period. The 100 mg dose demonstrated a significant effect versus placebo on DAS28-CRP<2.6 (23.1% vs 6.7%, p=0.016), all categories of the American College of Rheumatology (ACR) criteria (ACR20: 69.2% vs 40.0%, p=0.005; ACR50: 30.8% vs 12.0%, p=0.021; ACR70: 17.9% vs 4.0%, p=0.030), and the Health Assessment Questionnaire Disability Index (−0.48 vs −0.25, p=0.005). A biomarker-based disease activity score showed a dose-dependent decrease at week 12, indicating suppression of disease-related biological pathways. Adverse events were generally mild or moderate in intensity. No significant hypersensitivity reactions, serious or opportunistic infections, or changes in pulmonary parameters were observed.
Conclusions Mavrilimumab induced rapid clinically significant responses in RA subjects, suggesting that inhibiting the mononuclear phagocyte pathway may provide a novel therapeutic approach for RA.
- Rheumatoid Arthritis
- Treatment
- Disease Activity
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd to permit this article (if accepted) to be published in ARD and any other BMJPGL products and sublicences such use and exploit all subsidiary rights, as set out in our license.
Introduction
Despite the many treatments introduced for rheumatoid arthritis (RA), significant proportions of patients fail to achieve meaningful responses and are not adequately controlled.1 New therapies with novel mechanisms of action are still needed to address this unmet need. We postulate that direct modulation of macrophage function via granulocyte-macrophage colony-stimulating factor receptor (GM-CSFR) inhibition may provide a treatment option in RA. GM-CSF may play a central role in the pathogenesis of RA through the activation, differentiation and survival of neutrophils and macrophages.2 Macrophages promote synovitis via release of cytokines, chemokines, reactive oxygen and nitrogen intermediates, proteases and microparticles. The number of macrophages in synovial tissue is correlated with radiographic progression;3 reductions in CD68+ macrophages correlate with improvement in disease activity scores.4–6 RA patients exhibit increased GM-CSF levels in synovial fluid, plasma and synoviocytes,7–9 and recombinant GM-CSF has been reported to exacerbate disease in those patients.10–12
Mavrilimumab (CAM-3001) is a fully-human monoclonal antibody targeting the alpha subunit of GM-CSFR. In a phase 1 single ascending intravenous dose study in 32 subjects with RA, mavrilimumab showed a safety and tolerability profile supporting clinical development, and biological activity on acute phase reactants.13 This phase 2a study evaluated the efficacy and safety of subcutaneous (SC) mavrilimumab in subjects with moderate-to-severe active RA. This is the first study to investigate macrophage inhibition through direct blockage of GM-CSFRα as a novel therapeutic approach in RA.
Methods
Study design
This multicentre, randomised, double-blind, placebo-controlled study (EARTH Study; NCT01050998) evaluated the efficacy, safety and tolerability profile of ascending SC mavrilimumab doses in combination with stable methotrexate in subjects with moderate-to-severe active RA. Subjects were randomised between February 2010 and March 2011 at 53 centres across 10 Eastern European countries.
Subjects were randomised using an interactive voice response system in a 2:1 ratio (active:placebo) within each cohort and received, either 10, 30, 50, or 100 mg SC doses of mavrilimumab or placebo every other week for 12 weeks, followed by a 12-week follow-up period.
Doses and administration frequency were based on phase 1 data13 and pharmacokinetic-pharmacodynamic modelling.14 For Cohorts 1–3, dose escalation to the next cohort was based on a cumulative safety data review after ≥18 subjects completed day 29 dosing; for Cohort 4, six subjects received mavrilimumab 100 mg for the entire treatment period (12 weeks) and based on the safety review, randomisation was resumed.
Background stable non-steroidal anti-inflammatory drugs and oral corticosteroids (≤10 mg/day prednisolone or equivalent) were allowed.
The study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Guidance for Good Clinical Practice. Independent ethics committee approval was obtained. All subjects provided written informed consent. The protocol was jointly developed by the academic authors and the sponsor, MedImmune, who collected the data. Authors had full access to data and certify the veracity and completeness of the data and the data analysis.
Subjects
Subjects were 18−80 years with at least moderately active (Disease Activity Score (DAS)28-CRP ≥3.2)15 adult-onset RA of ≥3 months duration,16 despite methotrexate (7.5–25 mg/week) treatment for ≥12 weeks, and positive for anti-cyclic citrullinated peptide antibody (ACPA, >5 IU/ml; Axis Shield by ELISA) and/or rheumatoid factor (>14 IU/ml; Tina-Quant, Roche Diagnostics, Indianapolis, Indiana, USA and ELISA, Immco Diagnostics, Buffalo, New York, USA). All subjects received stable methotrexate (for ≥4 weeks) with supplemental folic acid ≥5 mg/week. Subjects previously treated with 1 biologic therapy for RA but discontinued for lack of efficacy were eligible for entry in the study. Subjects with symptomatic or uncontrolled lung disease, active infection, or at a high risk of infection were excluded.
Efficacy assessments
Efficacy assessments were performed at baseline and every 2 weeks during the 12-week treatment period and at 2 weeks, 1 month, and 3 months after the last dose. The primary endpoint was defined as the proportion of subjects achieving a reduction of ≥1.2 points from baseline in DAS28-CRP17 at week 12.
Secondary endpoints included time to onset of response and DAS28-CRP<2.6, improvements to the Health Assessment Questionnaire Disability Index (HAQ-DI)18 score, European League Against Rheumatism (EULAR), and American College of Rheumatology (ACR) response criteria.
Biomarker assessment
Twelve biomarkers (c-reactive protein (CRP), epidermal growth factor (EGF), Leptin, IL-6, MMP-1, MMP-3, Resistin, SAA, TNFR-I, VCAM-1, VEGF-A, YKL-40) were measured in serum samples from baseline and day 88 (Meso Scale Discovery MULTI-ARRAY platform). A multi-biomarker disease activity (MBDA) score was calculated for each sample by combining the concentrations of these biomarkers in the Vectra DA algorithm. The algorithm was trained and validated in previous studies using DAS28-CRP as a reference measure.19–21
Safety and tolerability assessments
Respiratory monitoring (chest x-ray, forced expiratory volume (FEV1), forced vital capacity (FVC), diffusing capacity of the lung for carbon monoxide (DLCO), and dyspnoea score) was performed at baseline and throughout the study because GM-CSF inhibition could affect alveolar macrophage function and surfactant homeostasis in the lung.22 ,23 The protocol mandated treatment discontinuation if pulmonary function deteriorated by >20% of baseline spirometry or DLCO values and for the changes to be reported as adverse events (AEs).
Serum surfactant D (SP-D) and KL-6 levels (ELISA; Sanko Junyaku Co, Japan)—established biomarkers for lung damage24–27—were measured at baseline and on days 29, 85, and 169. Other safety assessments included the incidence of AEs and serious AEs (SAEs) and routine laboratory testing.
Pharmacokinetics and immunogenicity
Serum mavrilimumab concentrations were measured from blood samples collected at the first and last doses, and at pre-designated timepoints using a validated electrochemiluminescence assay. The presence of anti-drug antibodies (ADA) was determined using a two-step approach: an electrochemiluminescence screening immunoassay and a confirmatory inhibition assay.
Statistical methods
Sample size calculations were based on the primary endpoint at week 12. We assumed a placebo response rate of 10%, a 15% drop-out rate, and used a two-sided significance level of 0.05, and a 2:1 (active:placebo) randomisation ratio. The total sample size of 216 subjects provided 86% power to detect a 20% difference in response rate between the combined mavrilimumab arms and placebo using Fisher's exact test. All response rates, including the primary endpoint, ACR20, ACR50, and ACR70, were analysed using Fisher's exact test. Changes from baseline in DAS28-CRP and HAQ-DI were analysed using a mixed-model repeated measures analysis with a covariate for baseline DAS28-CRP. The time-to-onset of response was analysed using a log-rank test.
All efficacy analyses were conducted on the intent-to-treat (ITT) population (all subjects randomised, regardless of whether they received treatment). Each analysis was conducted to compare the combined placebo and combined mavrilimumab groups, followed by comparison of the combined placebo group with each of the mavrilimumab dose cohorts. Analysis of safety data was carried out on the safety population (all subjects who received any dose of study medication).
For the primary endpoint as well as the other responder analyses, a non-responder imputation was used for subjects who withdrew from study treatment, changed the dose of background methotrexate, or received other RA medication. Other missing data points were imputed using last-observation-carried-forward methodology. No imputation was applied for the DAS28-CRP change from baseline analysis. Noncompartmental analysis was performed on individual pharmacokinetic (PK) data for the first and last doses. Biomarker data were analysed by Mann-Whitney U test and linear regression. Significance for individual biomarkers was evaluated by false discovery rate.28
Results
Of 427 subjects screened, 239 were subsequently randomised into the four cohorts. Of these, 233 were included in the ITT population, 216 completed the study, and 17 discontinued, three of which were due to AEs (figure 1). The treatment groups were generally balanced in terms of baseline and disease characteristics (table 1); the 50 mg group had a slightly higher proportion of female subjects and a lower mean dose of methotrexate.
Baseline and disease characteristics (Intent-to-treat population)

Patient disposition (ITT population).
Efficacy
Administration of mavrilimumab (all doses combined, n=158) was associated with a significantly higher proportion of subjects achieving a ≥1.2-point reduction in DAS28-CRP than placebo (n=75) at week 12 (55.7% vs 34.7%; p=0.003) (figure 2A). A significant difference in DAS28-CRP adjusted mean change from baseline compared with placebo was seen for the 50 mg (−0.38, 95% CI −0.67 to −0.08; p=0.013) and 100 mg (−0.44, 95% CI −0.74 to −0.14; p=0.004) cohorts as early as week 2 (figure 2B). DAS28-CRP<2.6 rates increased over time in all treated groups; the 100 mg dose demonstrated a significant effect (23.1% vs 6.7%; p=0.016) by week 12 and the number of subjects achieving DAS28-CRP<2.6 was still increasing at week 12 (figure 2A–C).
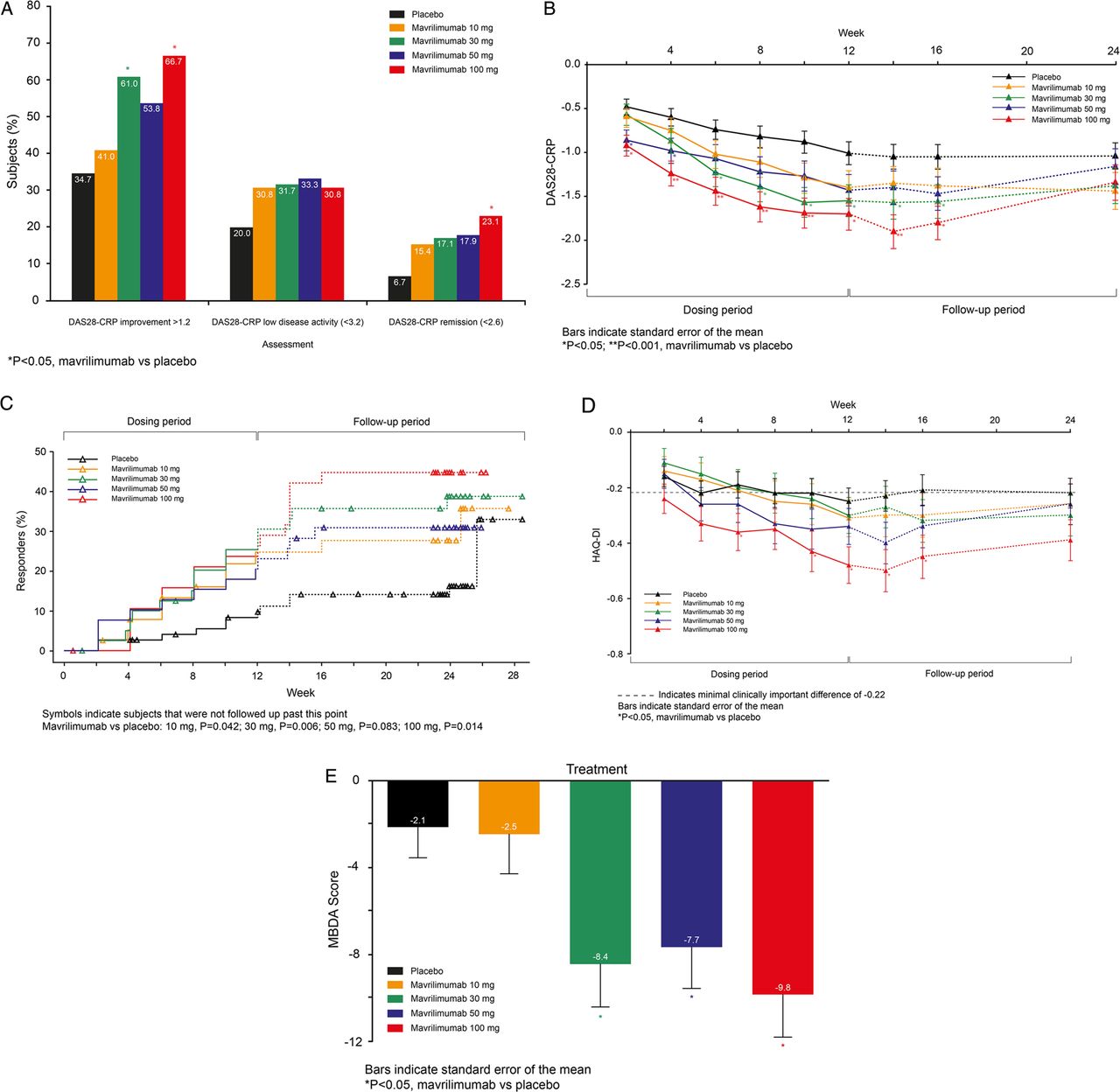
(A−E) Efficacy according to DAS-28 and Health Assessment Questionnaire Disability Index (HAQ-DI) assessments (ITT population). (A) DAS28-CRP responses by treatment group at week 12. (B) Mean DAS28-CRP by visit. (C) Time to onset of DAS28-CRP<2.6. (D) Adjusted mean HAQ-DI change from baseline by visit. (E) Changes in MBDA Score from baseline seen at week 12.

{kind=link}
{kind=link}
{kind=link}
Efficacy according to American College of Rheumatology (ACR) assessment (ITT population). (A) ACR responses by treatment group at week 12. (B) ACR20 responses by visit. (C) ACR50 responses by visit. (D) ACR70 responses by visit.
At the 100 mg dose, significant improvement in HAQ-DI versus placebo was observed from week 6 (−0.36 vs −0.19; p=0.041) through week 12 (−0.48 vs −0.25; p=0.005; figure 2D), with 74.4% mavrilimumab vs 48.0% placebo subjects reporting clinically significant improvements (≥0.25; p=0.009). The adjusted mean differences (95% CI) from baseline versus placebo were −0.17 (−0.33, −0.01) and −0.23 (−0.39, −0.07).
The largest treatment effect, as assessed by ACR criteria, was seen in the 100 mg dose at week 12 vs placebo (ACR20 69.2 vs 40.0%, p=0.005; ACR50 30.8 vs 12.0%, p=0.021; ACR70 17.9 vs 4.0%, p=0.030) (figure 3A) with a significant separation from placebo seen as early as week 4 (ACR20 53.8% vs 20.0% p<0.001) (figure 3B–D). A larger proportion of subjects receiving mavrilimumab showed a moderate or good EULAR response versus placebo (67.7 vs 50.7%; p=0.025). The highest proportion of moderate (46.2%) or good responders (30.8%) was seen in the 100 mg group.
Significant changes were seen from week 2 in CRP (p=0.004) and erythrocyte sedimentation rate (ESR) (p=0.005) in the mavrilimumab groups versus placebo, and in swollen and tender joint count from week 4 (p=0.002 and p=0.011, respectively) (see web only files).
Efficacy observed in the 100 mg group at week 12 was maintained through the follow-up to week 24, manifested both as a ≥1.2-point reduction in DAS28-CRP (59.0% vs 33.3% placebo; p=0.010) and ACR20 response (56.4% vs 30.7% placebo; p=0.009). This ‘post-treatment’ maintenance of response was not observed for DAS28-CRP<2.6, ACR50, and ACR70 (figures 2C and 3C,D).
Ninety-one per cent of subjects in the ITT population had sufficient sample available for biomarker analyses (placebo (n=65); mavrilimumab (n=146): 10 mg (n=35), 30 mg (n=37), 50 mg (n=38), and 100 mg (n=36)). At baseline, MBDA scores reflected the severe disease activity measured by DAS28-CRP. The MBDA score decreased more in the combined mavrilimumab-treated subjects versus placebo at week 12 (p=0.008) and decreases were dose-dependent (p<0.001) (figure 2E). There were also dose-dependent decreases in the constituent biomarkers CRP, IL-6, MMP-3, SAA, and YKL-40 (false discovery rate <0.05) (see web only files). Significant reductions in markers of systemic inflammation (CRP, SAA, IL-6) were observed within 1 month, with significant changes in YKL40 and MMP3 not being observed until week 12.
Safety and tolerability
Over the 24-week study period, 36 (45.6%) subjects receiving placebo and 91 (56.9%) receiving mavrilimumab experienced an AE (table 2).
Most frequent treatment-emergent AEs and most frequent treatment-related AEs (>1 subject in placebo or total mavrilimumab groups) (safety population)
Within the 12-week treatment period, 26 (32.9%) subjects receiving placebo and 73 (45.6%) subjects receiving mavrilimumab experienced an AE.
The protocol mandated that reductions in DLCO >20% of baseline be reported as AEs regardless of clinical significance. As such, these were the most frequently reported AEs (5 (6.3%) placebo and 19 (11.9%) mavrilimumab). Only two subjects (one placebo and one in the 10 mg cohort) discontinued because of DLCO reductions; no lung toxicities were detected as assessed by an independent pulmonologist. In the 30, 50, and 100 mg groups, there was no difference in DLCO event rate compared with placebo. Further, no clinically significant changes in serum SP-D and KL-6 were observed over the 3 months of treatment or 3 months follow-up across treatment groups (see web only files).
There were no clinically significant or persistent changes in lung function tests performed by spirometry. Nasopharyngitis and upper respiratory tract infections (all mild-to-moderate in severity) were the next most common events. Most AEs were mild or moderate in intensity.
There were no deaths during the study, and the frequency or severity of AEs was not dependent on mavrilimumab dose.
SAEs were reported in one subject receiving placebo (worsening of RA, which resulted in discontinuation) and four subjects receiving mavrilimumab; two in the 10 mg cohort, (intervertebral disc disorder and spontaneous abortion); and two in the 30 mg cohort, (fracture of the humerus and fracture of the patella). None of the SAEs was considered related to the study drug.
No instances of severe hypersensitivity, anaphylaxis, or serious injection site reactions (local or systemic) were reported during the treatment period. One (2.5%) subject in the 50 mg cohort experienced a mild rash, which was assessed as a hypersensitivity reaction that resolved spontaneously within 10 h.
Pharmacokinetics and Immunogenicity
Maximum serum concentration of mavrilimumab was observed approximately 3 days postdose, and PK steady-state was reached by day 57 in all dose groups. The PK exposure was more than dose-proportional. At the 100 mg dose, the terminal-phase PK half-life was ∼13 days.
During the study, 23 subjects developed detectable ADA (3 placebo and 20 treated). Generally, ADA titres were low and transient and did not impact the PK. High-titre ADAs were reported in 1 placebo (1.3%) subject and 10 mavrilimumab (6.3%; 3 at 10 mg, 4 at 30 mg, 1 at 50 mg, and 2 at 100 mg) subjects. Although the presence of ADA in these subjects was associated with reduced PK exposure, no correlation was observed with tolerability or tachyphylaxis.
Discussion
New therapeutic options capable of delivering a profound and rapid onset of action are desirable in RA because of the chronic progressive nature of the disease. Despite a variety of effective therapies currently available for the treatment of RA, many patients still fail to achieve clinical remission, show an inadequate response, or cannot maintain a response.1 ,29–31
Our study shows that mavrilimumab, the first investigational human monoclonal antibody to target GM-CSFRα, improves the signs and symptoms of RA in subjects with active disease despite stable methotrexate treatment. The study met its primary endpoint, and rapid and sustained improvement was demonstrated by DAS28-CRP, HAQ-DI, and ACR responses, particularly at the 100 mg dose. Significant improvements in DAS28-CRP were seen as early as week 2 at the two highest doses (50 and 100 mg), and in all dose groups, scores continued to improve throughout the 12-week treatment period. After treatment was discontinued at week 12, responses were sustained for ≥4 weeks; these diminished over the 12-week off-treatment period, but DAS28-CRP and ACR20 responses were maintained in the 100 mg group throughout this follow-up period. At week 24, the treatment response was maintained for HAQ-DI, swollen and tender joint counts, but there was no difference from placebo for the physician and patient global assessments, patient pain, CRP, and ESR. Of note is the observed placebo response rate, which was higher than assumed in the sample size calculation. Despite this, the primary endpoint was met and significant improvements were observed. Moreover, the differences in baseline characteristics in the 50 mg group were not found to account for the lower proportion of subjects achieving a ≥1.2-point reduction in DAS28-CRP or ACR20 in that treatment group.
Improvements in the MBDA composite scores complement and support the dose-dependent decrease in disease activity over the 12 weeks of treatment. Suppression of individual biomarkers within the panel suggests a beneficial modulation of pathophysiological pathways associated with RA including global inflammation (CRP, IL-6, SAA) and intra-articular pathology (MMP3, YKL-40).32–36 Changes in acute phase proteins and IL-6 occurred within 1 month, suggesting a direct effect on IL-6 production and consequently acute phase reactants. Chronic suppression of IL-6 with mavrilimumab may result in some of the beneficial mechanisms ascribed to IL-6 therapy in RA, such as suppressing Th17 cells, either indirectly37 or directly.38–40 The direct effects of GM-CSF inhibition on MMP3 and YKL-40 has not, to our knowledge, been described previously, but may indicate a beneficial effect on cartilage degradation. Further research is required to understand the effect of mavrilimumab on MMP3 biology in circulation and the joint.
Although we only evaluated the effects of mavrilimumab over 12 weeks, it was particularly encouraging that at the highest (100 mg) dose, 23.1% of subjects achieved DAS28-CRP<2.6 (placebo: 6.7%), and 17.9% showed an ACR70 response (placebo: 4.0%). Separation between the placebo and active groups was observed as early as week 4 for DAS28-CRP<2.6, suggesting a rapid onset of action even for this high-hurdle endpoint. The number of subjects achieving DAS28-CRP<2.6 and/or ACR70 response was still rising at 12 weeks, suggesting that peak efficacy may not have been achieved. It has been shown previously that response to some biologic treatments continues to increase over the first 24 weeks of treatment, and a significant proportion of partial responders and non-responders at week 12 may go on to achieve a clinical response with continued treatment.41
The safety profile was consistent with a previous phase 1 study of mavrilimumab in subjects with RA.13 Due to the link between GM-CSF and alveolar macrophage function and clearance of lung surfactant proteins, we carried out intensive pulmonary lung function tests as well as assays for biomarkers of lung damage such as SP-D and KL-6. At the study start, there was considerable variability in DLCO measurements, contributing to the increase in reported AEs in the 10 mg group. With greater experience with DLCO measurements, this AE rate was not observed in the subsequent cohorts. No meaningful differences were noted for SP-D and KL-6 between the active and placebo groups. Further, SP-D and KL-6 levels at baseline were representative of those described for healthy controls and RA patients with no interstitial lung disease.42–44 No serious or opportunistic infections or severe hypersensitivity reactions were reported in this patient population during the observation period.
The PK of mavrilimumab14 was nonlinear with dose due to GM-CSFR-mediated clearance. One placebo (1.3%) and 10 mavrilimumab-treated subjects (6.3%) developed ADA with a titre ≥4. Most incidences of ADA occurred in the lower dose groups (10 and 30 mg). Although these instances were associated with somewhat reduced PK exposure; there was no apparent correlation between ADA and hypersensitivities or reduced clinical responses. Longer-term studies will be necessary to understand the potential for mavrilimumab ADAs to affect the safety and/or efficacy profile of the molecule.
There are some important limitations to our study. It was a short-term, early phase study with a relatively small number of subjects in each treatment group. Because of potential safety concerns, doses were escalated sequentially, rather than by adopting the standard parallel-group design.
Despite these limitations, our study suggests that targeting the alpha subunit of the GM-CSFR may be a novel approach to RA treatment. Mavrilimumab, especially at the higher doses, appears to produce rapid and clinically meaningful effects across a number of disease activity parameters with no unexpected safety concerns. This hypothesis will be evaluated in future larger, appropriately powered clinical studies.
Acknowledgments
The authors wish to acknowledge Geraldine Grove, Didier Saurigny, Bente Larson, and Mark Hopton for their support in the conduct and interpretation of the study. Editorial assistance was provided by Eleanor Steele, BSc, from QXV Communications, Macclesfield, UK. The authors thank Ruth Pereira, PhD, for her critical review of the manuscript and her valuable comments.
The authors would like to thank the following EARTH study investigators for their valuable contribution to the study: Bulgaria: Anastas Batalov, Ivan Goranov, Vladimir Kadinov, Rumen Stoilov, Daniela Yaneva; Czech Republic: Jiri Vencovsky, Ladislav Bortlik, Petr Vitek, Dagmar Galatikova, Zuzana Urbanova, Zuzana Stejfova; Estonia: Eevi Parsik, Jaak Talli, Ivo Valter; Hungary: Ildiko Kiss, Janos Bartalos, Istvan Szombati, Marianne Szongoth; Latvia: Helena Mikazane, Samite Saleniece, Daina Saulite-Kandevica; Lithuania: Sigitas Stonkus, Algirdas Venalis; Poland: Dariusz Chudzik, Stefan Daniluk, Przemyslaw Kotyla, Iwona Swierzewska-Olech, Leszek Szczepanski, Malgorzata Szymanska, Piotr Leszczynski, Wlodzimierz Samborski, Maria Stopinska- Polaszewska, Grazyna Krzyzanowska; Romania: Florin Radulescu, Marian Sandu; Russia: Olga Ershova, Anna Galustyan, Alexander Gordienko, Nikolay Korshunov, Galina Matsievskaia, Ildar Salikhov, Valeriy Shirinskiy, Elena Zonova, Olga Barbarash, Galina Chumakova, Andrey Dorokhov; Ukraine: Kateryna Amosova, Oleksandr Dyadyk, Mykola Vatutin, Volodymyr Kovalenko, Vladislav Povoroznyuk, Semen Ter-Vartanyan, Grigoriy Ignatenko
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Lay summary
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Handling editor Tore K Kvien
-
Funding The study was sponsored by MedImmune Ltd.
-
Competing interests Gerd Burmester is a consultant for MedImmune, Abbott, BMS, MSD, Pfizer, Roche, and UCB; has received travel reimbursement for meetings for the study from MedImmune; and has grants from and is a speaker for Abbott, BMS, MSD, Pfizer, Roche, and UCB. Michael Weinblatt is a consultant for Abbott, Amgen, Astellas, AstraZeneca, Biogen/Idec, BMS, Crescendo Biosciences, Hoffman La Roche, Janssen (Centocor), Lilly, MedImmune, Merck, Pfizer, UCB, and Vertex. He has received funding for travel expenses to attend consulting meetings for the companies listed above and has grants from Biogen/Idec, Crescendo Bioscience, and MedImmune. Iain McInnes is a consultant for MedImmune, AstraZeneca, Boehringer Ingelheim, Crescendo Bioscience, Johnson & Johnson, NovoNordisk, Pfizer, Roche, and UCB has grants from AstraZeneca, BMS, MSD, Pfizer, Roche, UCB; and is a speaker for BMS, MSD, Pfizer, Roche, and UCB. Duncan Porter has received fees for participation in the Mavrilimumab Development Advisory Board; is a consultant for BMS, Pfizer, and Roche; has grants from Pfizer and Roche; is a speaker for Abbott; and has received travel reimbursement from Abbott, BMS, Roche, and UCB. Olga Barbarash, Mykola Vatutin, and Istvan Szombati have nothing to disclose. Guy Cavet is an employee of and has stock options from Crescendo Bioscience. Ehsanollah Esfandieri, Chris Kane, and Fabio Magrini were employees of MedImmune while the study was being conducted; Fabio Magrini owned stock in AstraZeneca. Matt Sleeman and Alex Godwood are employees of MedImmune and own stock in AstraZeneca. Bing Wang is an employee of MedImmune.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Basic and translational research
- Clinical and epidemiological research
- Clinical and epidemiological research