Article Text

Abstract
Purpose To evaluate the efficacy and safety of adalimumab in patients with non-radiographic axial spondyloarthritis (nr-axSpA).
Methods Patients fulfilled Assessment of Spondyloarthritis international Society (ASAS) criteria for axial spondyloarthritis, had a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of ≥ 4, total back pain score of ≥ 4 (10 cm visual analogue scale) and inadequate response, intolerance or contraindication to non-steroidal anti-inflammatory drugs (NSAIDs); patients fulfilling modified New York criteria for ankylosing spondylitis were excluded. Patients were randomised to adalimumab (N=91) or placebo (N=94). The primary endpoint was the percentage of patients achieving ASAS40 at week 12. Efficacy assessments included BASDAI and Ankylosing Spondylitis Disease Activity Score (ASDAS). MRI was performed at baseline and week 12 and scored using the Spondyloarthritis Research Consortium of Canada (SPARCC) index.
Results Significantly more patients in the adalimumab group achieved ASAS40 at week 12 compared with patients in the placebo group (36% vs 15%, p<0.001). Significant clinical improvements based on other ASAS responses, ASDAS and BASDAI were also detected at week 12 with adalimumab treatment, as were improvements in quality of life measures. Inflammation in the spine and sacroiliac joints on MRI significantly decreased after 12 weeks of adalimumab treatment. Shorter disease duration, younger age, elevated baseline C-reactive protein or higher SPARCC MRI sacroiliac joint scores were associated with better week 12 responses to adalimumab. The safety profile was consistent with what is known for adalimumab in ankylosing spondylitis and other diseases.
Conclusions In patients with nr-axSpA, adalimumab treatment resulted in effective control of disease activity, decreased inflammation and improved quality of life compared with placebo. Results from ABILITY-1 suggest that adalimumab has a positive benefit–risk profile in active nr-axSpA patients with inadequate response to NSAIDs.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Axial spondyloarthritis may be split into two categories—ankylosing spondylitis (AS) and non-radiographic axial spondyloarthritis (nr-axSpA)—by the 1984 modified New York criteria which require the presence of sacroiliitis on plain x-rays for the classification of AS.1 ,2 With the availability of MRI, the presence of inflammation in the axial skeleton in the absence of radiographic changes can now be assessed.2–4 The Assessment of Spondyloarthritis international Society (ASAS) has validated new classification criteria for patients with axial SpA5 ,6 to establish standards that apply to patients with or without radiographic sacroiliitis by including both plain x-rays and MRI as imaging modalities.5 Although these criteria identify both AS and nr-axSpA patients, they also allow the conduct of clinical trials for the treatment of nr-axSpA which is considered an unmet medical need.5
Registry and clinical trial data indicate that patients with AS and those with nr-axSpA have comparable clinical manifestations and burden of disease, requiring treatment irrespective of the presence of radiographic damage.7–9 Non-steroidal anti-inflammatory drugs (NSAIDs) are considered first-line therapy for all patients with axial SpA.10 ,11 Traditional disease-modifying antirheumatic drugs (DMARDs) such as methotrexate and sulfasalazine are not effective for the treatment of axial SpA.11–14 Anti-tumour necrosis factor (TNF) agents are approved therapies for patients with AS who continue to have active disease despite NSAIDs. However, for patients with nr-axSpA there is currently no alternative treatment to NSAIDs.
New treatment recommendations for patients with axial SpA, including nr-axSpA, identify anti-TNF agents as options in patients who fail NSAIDs;11 however, large phase III trials for this population have been lacking. Previous clinical trials conducted prior to the availability of the ASAS axial SpA criteria employed variable definitions of nr-axSpA and were limited by small sample sizes; nonetheless, these studies provided data showing that anti-TNF therapy is also effective in nr-axSpA.9 ,15
The present study was developed to further evaluate the efficacy and safety of adalimumab in the treatment of patients with nr-axSpA who have active disease despite treatment with NSAIDs. This represents the first randomised controlled clinical trial to use the ASAS axial SpA criteria in classifying patients with nr-axSpA.
Methods
Patients
Eligible patients were ≥18 years of age and fulfilled ASAS classification criteria for axial SpA6 without meeting modified New York criteria for AS.1 Patients must have had active disease, exhibited by a total back pain score of ≥4 on a 0–10 cm visual analogue scale (VAS) (≥40 on a 0–100 mm VAS) and a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of ≥4. They must also have responded inadequately or been intolerant to one or more NSAIDs, or had a contraindication to NSAIDs based on the clinical judgment of the investigator. Patients with previous or current diagnoses of psoriasis or psoriatic arthritis or a history of inflammatory arthritis of a different aetiology were excluded. Previous exposure to biological agents was not permitted.
Study design
ABILITY-1 (NCT00939003) was initiated in August 2009 and is an ongoing phase III randomised placebo-controlled double-blind trial conducted at 37 centres in Australia, Belgium, Canada, Czech Republic, France, Germany, Spain, The Netherlands, the UK and the USA. It was conducted in accordance with International Conference on Harmonization good clinical practices and the Declaration of Helsinki. Approval of an institutional ethics review board and voluntary written informed patient consent were obtained prior to study procedures.
Eligible patients were centrally randomised using an interactive voice response system 1:1 to receive subcutaneous injections of adalimumab (40 mg every other week) or matching placebo for 12 weeks during the double-blind period. Efficacy and safety were assessed at weeks 2, 4, 8 and 12. Patients who completed the double-blind period were eligible to receive open-label adalimumab for up to an additional 144 weeks.
Patients could enter the study on concomitant NSAIDs, prednisone (≤10 mg per day), methotrexate (MTX, ≤25 mg per week), sulfasalazine (SSZ, ≤3 g per day) and/or hydroxychloroquine (≤400 mg per day) or azathioprine (≤150 mg per day, but not concomitant with any other DMARD) if the doses met pre-specified stability requirements prior to randomisation and remained stable during the first 24 weeks except as medically required due to an adverse event (AE).
Efficacy endpoints
Primary efficacy endpoint
The primary efficacy endpoint was the proportion of patients who achieved an ASAS40 response at week 12. An ASAS40 response was defined as a ≥40% improvement and an absolute improvement from baseline of ≥2 units (range 0–10) in ≥3 of the following four domains: Patient Global Assessment of Disease Activity (0–10 cm VAS), pain (total back pain, 0–10 cm VAS), function (Bath Ankylosing Spondylitis Functional Index (BASFI), 0–10 cm VAS16) and inflammation/morning stiffness (mean score of items 5 and 6 of the BASDAI (0–10 cm VAS)) without any worsening in the remaining domain.17
Secondary efficacy endpoints
Secondary efficacy variables analysed at week 12 included: ASAS20,18 ASAS partial remission (ASAS PR),18 ASAS5/6,17 BASDAI (0–10 cm VAS), BASDAI50, Ankylosing Spondylitis Disease Activity Score (ASDAS),19 ,20 ASDAS clinically important improvement (ASDAS CII, decrease from baseline ≥1.1), ASDAS major improvement (ASDAS MI, decrease from baseline ≥2.0), ASDAS inactive disease (ASDAS ID, score <1.3),21 Maastricht Ankylosing Spondylitis Enthesitis Score (MASES, 0–13),22 linear Bath Ankylosing Spondylitis Metrology index (BASMIlin, 0–10),23 36-Item Short Form V.2 Health Survey (SF-36), Health Assessment Questionnaire modified for Spondyloarthropathies (HAQ-S)24 and Spondyloarthritis Research Consortium of Canada (SPARCC) MRI scores for the sacroiliac (SI) joints (0–72)25 and the spine (0–108).26 MRI films were scored by two independent central readers who were blinded to time point and sequence. Average scores of the readers were used.
Safety assessments
Treatment-emergent AEs were defined as AEs that began or worsened after the first dose of study medication through 70 days after the last dose.
Statistical analysis
Efficacy variables were analysed for all randomised patients who received at least one dose of blinded study medication, but excluding seven patients from one site due to investigator non-compliance. The safety population consisted of all patients who received at least one dose of study medication.
A target sample size of 194 patients (97 placebo and 97 adalimumab) was calculated to provide approximately 90% statistical power to detect a 20% difference in ASAS40 response rates between the treatment groups, based on a two-sided χ2 test with a significance level of 0.05.
For categorical variables, patients with missing data at week 12 were considered to be non-responders using non-responder imputation (NRI). Last observation carried forward imputed values were used for continuous variables. Analysis of covariance (ANCOVA) adjusting for the baseline score was used to compare change from baseline at week 12 between adalimumab and placebo treatment groups. VAS data were collected on 0–100 mm scales and reported as 0–10 cm data for consistency.
To evaluate the impact of baseline demographics and disease conditions on the primary efficacy endpoint, ASAS40 response at week 12 was summarised by subgroups of sex (male, female), race (white, non-white), age (<40, ≥40 years), weight (<70, ≥70 kg), symptom duration (<5, ≥5 years), baseline C-reactive protein (CRP) (normal, elevated), concomitant baseline NSAID use (yes, no) or DMARD use (yes, no), history of inflammatory bowel disease (yes, no) or uveitis (yes, no), baseline HLA-B27 status (positive, negative), past or current MRI evidence of inflammation of the SI joints according to the local radiologist/rheumatologist (positive, negative) and baseline SPARCC SI joint score (<2, ≥2). For subgroup analyses, a logistic model was used to assess treatment and subgroup interaction, with a significant interaction defined as p≤0.10.
AEs were summarised as the number and percentage of patients experiencing AEs using Medical Dictionary for Regulatory Activities (MedDRA, V.13.1) system organ classes and preferred terms.
Results
Patients
There were 192 patients randomised into the study. Due to investigator non-compliance identified at a single site, seven patients (three placebo, four adalimumab) were excluded from all efficacy analyses but were included in safety analyses. Of the remaining 185 patients, 94 received placebo and 91 received adalimumab (figure 1). Through the 12-week double-blind period, one patient from each treatment group discontinued due to AEs. One placebo patient and three adalimumab patients discontinued for other reasons, which included pregnancy, lack of efficacy or violation of entry criteria.

ABILITY-1 patient disposition. a ‘Other’ reasons for discontinuation included lack of efficacy, pregnancy or violation of entry criteria. One subject in the adalimumab group had a week 12 visit but discontinued at that visit due to a positive pregnancy test and did not receive study drug at the week 12 visit.
Patient demographics were comparable between the treatment groups (table 1), with no significant differences noted between adalimumab and placebo patients. Over half of the patients (55%) were women and the mean age across both groups was 38 years. Despite a mean 10-year history of symptoms, the average time since diagnosis was only approximately 3 years prior to study entry.
Baseline demographics and disease characteristics for the full analysis set and subgroups of patients fulfilling the imaging or clinical arms of the Assessment of Spondyloarthritis international Society axial SpA criteria
The most common SpA features included as components of the ASAS axial SpA criteria noted in the study population were inflammatory back pain, HLA-B27 positivity and a good prior response to NSAIDs. Despite the absence of radiographic sacroiliitis, patients with nr-axSpA had high levels of disease activity as measured by the BASDAI and ASDAS. Furthermore, there were moderate levels of functional impairment and reduction in quality of life despite low baseline BASMIlin scores, indicating minimal impairment of spinal mobility. The low mean MASES score at baseline also indicated that there was limited involvement or inflammation of entheses in the overall study population, although 72% (133/185) of patients had MASES>0 at baseline (74%, 70/94 placebo; 69%, 63/91 adalimumab).
With the exception of HLA-B27 status, there were no striking differences between patients who fulfilled the imaging arm (positive MRI sacroiliitis according to the local radiologist/rheumatologist) and those who fulfilled the clinical arm (HLA-B27 positive/MRI negative) of the ASAS axial SpA criteria (table 1). However, more patients who fulfilled the clinical arm of the ASAS criteria had a family history of SpA. The SPARCC MRI scores for SI joints were also lower in patients who fulfilled the clinical arm, but the scores for the spine were comparable across groups.
Efficacy
A significantly greater percentage of nr-axSpA patients treated with adalimumab achieved the primary endpoint of ASAS40 response at week 12 (33/91, 36%) compared with patients treated with placebo (14/94, 15%; p<0.001, NRI; figure 2A). Based on subgroup interaction analyses, symptom duration, age and baseline CRP status showed significant interactions with treatment on ASAS40 response (p=0.02, p=0.05 and p=0.03, respectively; NRI). There was a greater treatment effect with adalimumab among patients with symptom duration <5 years (figure 2B), those whose age was <40 years (figure 2C) and patients who had elevated CRP levels at baseline (figure 2D). HLA-B27 status did not demonstrate a significant treatment interaction with adalimumab (p = 0.42; NRI; figure 2E). Patients achieved similar ASAS40 responses with adalimumab regardless of whether or not they had past or present sacroiliitis on MRI according to the assessment of the local radiologist/rheumatologist (p=0.65; NRI). Sensitivity analyses to further explore the influence of MRI inflammation in the SI joints were conducted using centrally-read SPARCC scores. Stratifying patients using the operational classification of positive or negative SI joint MRI inflammation based on SPARCC scores ≥2 or <2 at baseline showed a numerically greater treatment effect of adalimumab in patients with SI joint scores ≥2, but the interaction was not statistically significant (p=0.31; figure 2F). However, a significant interaction with treatment was observed based on logistic regression between continuous SPARCC SI joint scores at baseline and ASAS40 response at week 12 (p=0.046, NRI; supplementary online Figure S1). Finally, patients with either a positive MRI (SPARCC score ≥2 for either the SI joints or spine) or an elevated CRP at baseline demonstrated a greater response to adalimumab compared with placebo (41% (28/69) adalimumab vs 14% (10/73) placebo), in contrast to patients who had negative MRI of the SI joints and spine and a normal CRP at baseline (23% (5/22) adalimumab vs 20% (4/20) placebo), although the interaction was not statistically significant (p=0.13, NRI). Sex, race, weight, concomitant NSAID or DMARD use and history of inflammatory bowel disease or uveitis did not significantly affect the response to adalimumab treatment.

Percentage of patients achieving Assessment of Spondyloarthritis international Society 40 response at week 12. (A) Full analysis set; *p<0.001 for comparison of treatment response between adalimumab versus placebo. (B) Patients with symptom duration <5 years or ≥5 years. (C) Patients with age <40 years or ≥40 years. (D) Patients with or without elevated C-reactive protein (CRP) at baseline. (E) Patients with presence or absence of HLA-B27. (E) Patients with SPARCC SI joint score <2 or ≥2 at baseline. Non-responder imputation.
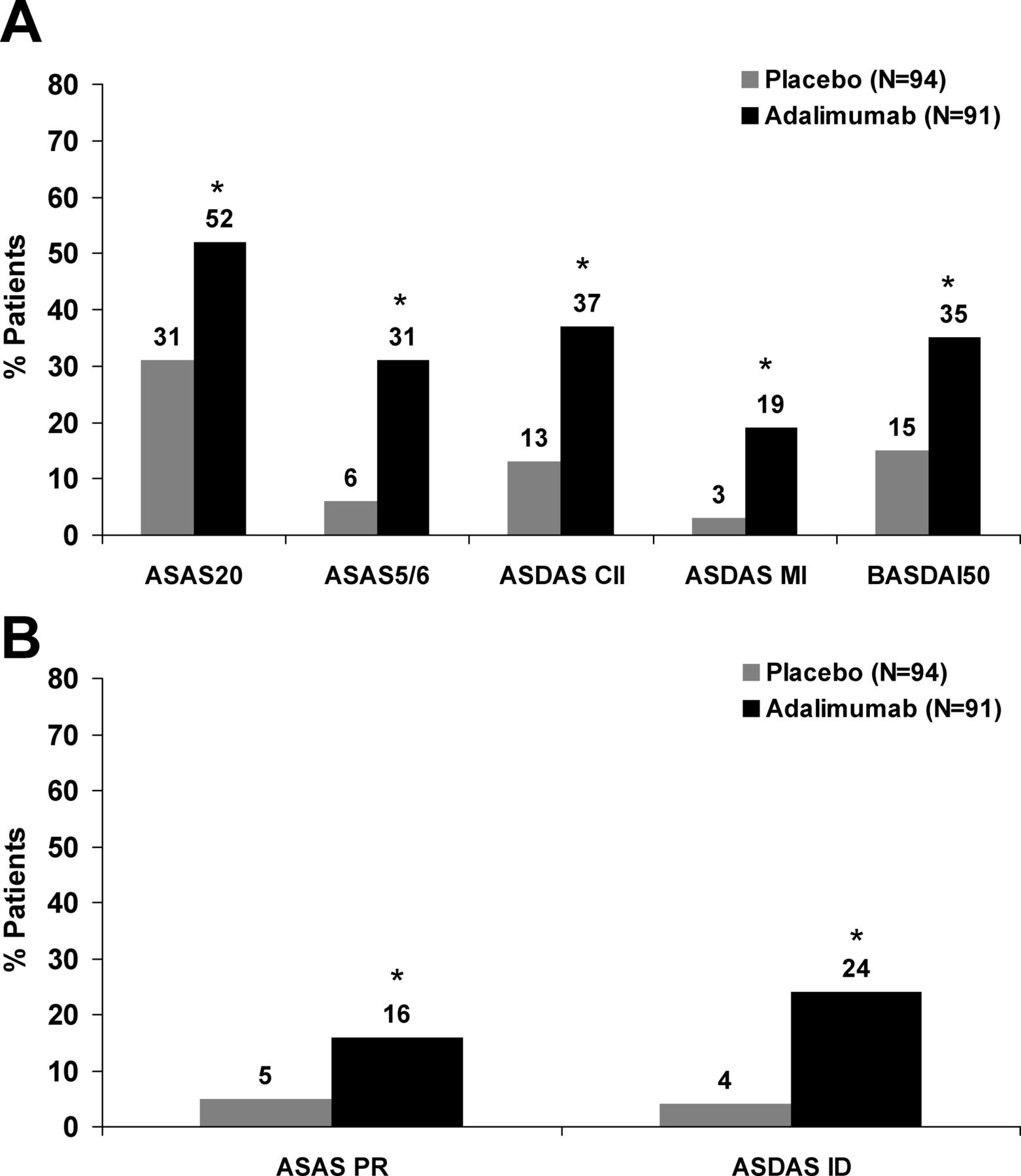
Adalimumab treatment resulted in significantly higher percentages of nr-axSpA patients achieving clinical improvements as measured by ASAS, ASDAS and BASDAI response criteria (figure 3A). Significantly higher percentages of adalimumab-treated patients also achieved states of disease remission (figure 3B), with approximately one in four patients achieving ASDAS ID after 12 weeks of treatment with adalimumab.

{kind=link}
{kind=link}
{kind=link}
(A) Percentage of patients with clinical response. (B) Percentage of patients with disease remission. Non-responder imputation. *p≤0.001 for comparison of treatment response between adalimumab versus placebo except Assessment of Spondyloarthritis international Society 20 (ASAS20) (p=0.004) and ASAS PR (p=0.01). ASDAS CII, clinically important improvement; ASDAS MI, major improvement; ASAS PR, partial remission; ASDAS ID, inactive disease.
Adalimumab treatment was associated with statistically significant improvements in disease activity of nr-axSpA patients whether measured by composite indices (ie, BASDAI, ASDAS) or by individual objective measures of inflammation (ie, CRP, MRI scores) when compared with placebo (table 2). In particular, there were significant decreases in each of the individual components of the ASAS40 response criteria except for BASFI. Although there was a numerically greater improvement in BASFI with adalimumab than with placebo, the difference was not statistically significant. Based on the BASFI component of the ASAS partial remission criteria, 33% of patients (25/75) had BASFI <2 at week 12 in the adalimumab group compared with 11% (9/79) in the placebo group (p=0.001) among patients with BASFI ≥2 at baseline. Minor decreases in the BASMIlin and the MASES score were also observed but were not significantly different between treatment groups. Significant improvements in physical function and quality of life were noted with adalimumab, as determined by the HAQ-S and SF36 physical component summary scores.
Clinical, functional and imaging endpoints at week 12
Safety
During the double-blind period, similar proportions of patients in the two treatment groups experienced any AE or an infectious AE (table 3). The most common events were nausea (8.2%), diarrhoea (7.2%) and upper respiratory tract infection (4.1%) among patients in the placebo group and nasopharyngitis (11.6%), nausea (7.4%) and headache (6.3%) in the adalimumab group. There were few serious AEs. A patient in the placebo group had a serious AE with symptoms of nausea, vomiting, chills, fever and dizziness. Three serious AEs were reported in the adalimumab group: breast dysplasia, induced abortion and acute hepatitis. The acute hepatitis was reported by the investigator as probably due to isoniazid for tuberculosis prophylaxis as the baseline visit laboratory tests prior to the first dose of study drug but after starting isoniazid showed elevated liver function tests. The case of acute hepatitis was one of four hepatic-related AEs in the adalimumab group; all other hepatic-related AEs were isolated liver function test abnormalities that were not associated with a definite diagnosis or liver pathology. There were no malignancies, opportunistic infections, tuberculosis, lupus-like syndrome, demyelinating disease or deaths through week 12.
Number and percentage of patients with adverse events (AEs) during the 12-week double-blind period
Discussion
This study is the first clinical trial to incorporate the ASAS axial SpA criteria in classifying nr-axSpA patients and the largest randomised controlled trial of an anti-TNF therapy in this population. Results from this study provide important insights into the characteristics of patients with nr-axSpA and the potential benefits of adalimumab therapy. First, the level of disease activity of nr-axSpA patients enrolled in this study was similar to those of AS patients who participated in randomised controlled trials of anti-TNF therapies. Second, significant clinical improvement was observed in nr-axSpA patients treated with adalimumab compared with placebo after just 12 weeks of therapy. Third, consistent with previous reports, the clinical response to adalimumab treatment in nr-axSpA patients was greater in patients with shorter symptom duration, younger age or presence of elevated CRP at baseline.
The nr-axSpA population enrolled in this study was younger and had a greater proportion of women compared with AS populations in randomised controlled trials of anti-TNF therapies.8 ,27 ,28 However, these observations are consistent with what has been reported for other nr-axSpA cohorts, including the patient population that was evaluated to validate the ASAS axial SpA criteria.6 ,7 ,9 ,29 There was an average delay of 7 years between onset of symptoms to diagnosis in nr-axSpA patients in this study, which highlights the need for better ways to identify these patients. Despite the absence of radiographic sacroiliitis and the above differences in demographics compared with AS patients, patients with nr-axSpA who continued to have symptoms of active disease despite NSAIDs had comparable levels of disease activity to that of patients with AS according to BASDAI, ASDAS and total back pain.8 ,9 ,27 ,28 However, a smaller proportion of patients with nr-axSpA had elevated CRP at baseline compared with AS patients, and mean MRI SPARCC scores for the SI joints and spine at baseline were lower than previous reports in patients with AS.8 ,27 ,28 ,30
The study met its primary endpoint with 36% of patients in the adalimumab group achieving ASAS40 response at week 12 compared with 15% in the placebo group. ASAS40 is a more stringent measure of response than ASAS20 typically used in AS trials. Treatment effect with adalimumab was also significant when other composite measures were used (ie, ASAS20, ASAS 5/6, BASDAI50, ASDAS). More importantly, clinical remission was achieved by more patients in the adalimumab arm than in the placebo arm, whether defined by ASAS PR or ASDAS ID. The clinical efficacy of adalimumab was further supported by significant improvements in objective measures of inflammation (ie, CRP and the SPARCC MRI scores for both SI joints and spine). Although improvements in functionality and quality of life were noted based on the SF36 physical component summary score and HAQ-S, improvement in BASFI and BASMIlin did not meet statistical significance. This could be attributed in part to the relatively low baseline BASFI and BASMIlin scores. These results confirm previous findings from smaller trials of anti-TNF agents in nr-axSpA. In a randomised controlled trial of 46 patients with nr-axSpA, adalimumab therapy resulted in significantly better ASAS40 responses at week 12 compared with placebo.9 Likewise, in 40 patients with inflammatory back pain for 3 months to 3 years, HLA-B27 positivity and MRI evidence of sacroiliitis, patients receiving infliximab had significantly greater reduction in BASDAI and MRI scores than those on placebo at week 16.15
Subgroup analyses of the interaction between certain baseline characteristics and treatment showed that patients whose symptom duration was <5 years, age <40 or baseline CRP was elevated were more likely to achieve ASAS40 at week 12 with adalimumab. Similar observations have been reported in established AS 31–33 and were noted in a randomised controlled trial of adalimumab in 46 patients with active axial SpA without radiographically-defined sacroiliitis: patients with disease duration ≤3 years, age ≤30 or CRP>6 mg/l at baseline had a greater probability of achieving ASAS40 or BASDAI50 at week 52.9 These findings further emphasise the need to diagnose patients earlier. No other baseline variables evaluated had an impact on treatment response, including HLA-B27 status and past or present sacroiliitis on MRI according to the ASAS criteria, suggesting that adalimumab is a potential treatment option for nr-axSpA patients regardless of whether they fulfil the imaging or clinical arm of the ASAS axial SpA criteria. However, patients with active inflammation of the SI joints based on a SPARCC score ≥2 at baseline showed a numerically (although non-significant) higher response, and increasing baseline SPARCC SI joint scores were associated with a greater likelihood of clinical response. These findings suggest that the greater the extent of SI joint inflammation, the more likely a patient may benefit from adalimumab treatment. There was also a trend indicating better responses in patients with either inflammation of the SI joints or spine or an elevated CRP at baseline. Thus, objective evidence of active inflammation at baseline, such as presence of a positive MRI or an elevated CRP level, seems to be a good predictor of treatment response to adalimumab.
Adalimumab was well tolerated during the double-blind period of this study. There were no notable imbalances in the occurrence of AEs between treatment groups. Safety data are consistent with what is known about the safety profile of adalimumab in AS and other immune-mediated diseases.8 ,34
Limitations of this study include the duration of the double-blind period, which does not allow for longer term comparison of the efficacy of adalimumab therapy with placebo in nr-axSpA patients who continue to have active disease despite NSAIDs. This study was not designed to evaluate if adalimumab therapy can prevent progression from nr-axSpA to AS. The trial was also not powered for subgroup analyses, which were further limited by uneven distribution of patients in certain subgroups (eg, HLA-B27 status). In addition, the outcome measures used in this study were validated for AS and have not been specifically developed and validated for a nr-axSpA population. As patients with nr-axSpA and those with AS are part of the same disease spectrum of axial SpA and have similar disease manifestations, the use of previously developed outcome measures for AS in this study of nr-axSpA patients was deemed to be appropriate.
In summary, 12 weeks of adalimumab therapy in patients with nr-axSpA resulted in significant clinical improvements compared with placebo, providing additional evidence that adalimumab controls inflammation with a similar safety profile across a range of spondyloarthritides. Efficacy and safety results from this study suggest that adalimumab is an appropriate treatment option for active nr-axSpA patients who fail NSAIDs, especially those with objective evidence of inflammation. Longer term data would provide information on the optimal use of adalimumab in patients with nr-axSpA.
Acknowledgments
Suchitrita Sarkar of Abbott provided support for statistical analyses and Elaine M Smith of Abbott assisted with drafting and revising the manuscript with direction from the authors.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure 1
Footnotes
-
Contributors JS was the principal investigator for the study. ALP and VA designed the study and statistical analysis plan. JS, DvdH, MD, PJM, WPM and MAB participated in collection of the data. VA supervised the statistical analyses. All authors contributed to interpretation of the data and critically reviewed and approved the final version of the manuscript for publication in accordance with the International Committee of Medical Journal Editors authorship criteria.
-
Funding The study was sponsored by Abbott Laboratories (NCT00939003).
-
Competing interests JS has received research grants, speaker's fees or consulting fees from Abbott, Merck, Pfizer and UCB. DvdH has received consulting fees and/or research grants from Abbott, Amgen, AstraZeneca, BMS, Centocor, Chugai, Eli-Lilly, GSK, Merck, Novartis, Otsuka, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, UCB and Wyeth. MD has received research grants and has participated at advisory boards and/or symposia in the field of spondyloarthritis organised by Abbott, BMS, Merck, Novartis, Pfizer, Sanofi-Aventis and UCB. PJM has received research grants, consultant fees, and speaker's bureau from Abbott, Amgen, BiogenIdec, BMS, Genentech, Janssen, Lilly, Pfizer, and UCB,and research grants and consultant fees from Celgene, Novartis, and Roche. WPM has received research grants and/or speakers’ or consulting fees from Abbott, BMS, Eli-Lilly, Janssen, Pfizer and UCB. MAB has received research grants, speaker's fees or consulting fees from Abbott, Eli-Lilly, Jansen-Cilag, Merck, Pfizer, Procter and Gamble, Schering-Plough and Wyeth. VA and ALP are Abbott employees and may hold Abbott stock or options.
-
Ethics approval Ethics approval was obtained from the appropriate local ethics committee at each study site.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Linked Articles
- Editorial
- Clinical and epidemiological research