Article Text

Abstract
Objectives To identify a new disease-modifying osteoarthritis drug (DMOAD) candidate that can effectively repair cartilage by promoting chondrogenic differentiation and halt osteoarthritis (OA) progression by suppressing aberrant hypertrophy.
Methods We screened 2500 natural and synthetic small compounds for chondrogenic agents via four steps using the Col2GFP-ATDC5 system and identified a small thienoindazole derivative compound, TD-198946, as a novel DMOAD candidate. We tested its efficacy as a DMOAD via intra-articular injections directly into the joint space in a surgically-induced mouse model of OA both at the onset (prevention model) and 4 weeks after (repair model) OA induction. The downstream molecules were screened by microarray analysis. We further investigated the mechanism of the drug action and its molecular target using in vitro and in vivo assays.
Results TD-198946 strongly induced chondrogenic differentiation without promoting hypertrophy in cell and metatarsal organ cultures. When administered directly into the joint space, TD-198946 successfully prevented and repaired degeneration of the articular cartilage. TD-198946 exerted its effect through the regulation of Runx1 expression, which was downregulated in both mouse and human OA cartilage compared with normal tissue.
Conclusions Our data suggest that TD-198946 is a novel class of DMOAD candidate, and that targeting Runx1 will provide a promising new approach in the development of disease-modifying drugs against OA.
- Chondrocytes
- Knee Osteoarthritis
- Osteoarthritis
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA), the most common degenerative disorder of the joints, affects nearly half the elderly population worldwide and represents an enormous socioeconomic challenge.1 ,2 The disease results from an imbalance in the dynamic equilibrium between the breakdown and repair of the joint tissues, often in situations where mechanical loading exceeds that which can be tolerated by the joint. However, the exact pathogenesis of OA is still a subject of debate and research.
During development, mesenchymal progenitors differentiate into chondrocytes and produce cartilage-specific matrix proteins such as type II collagen (Col2a1) and glycosaminoglycans (GAGs) such as aggrecan (Acan)3 under the influence of the master regulators of chondrogenic differentiation—the sex-determining region Y-type high mobility group box protein Sox9 and its cofactors Sox5 and Sox6. Thereafter, most chondrocytes undergo hypertrophy, as characterised by the secretion of type X collagen (Col10a1) which triggers cartilage degradation and mineralisation via endochondral ossification. Runx family proteins, runt-related transcription factors, play crucial roles in this process.4 Runx1 has been shown to be an important factor for early chondrogenic differentiation.5 Conversely, chondrocytes at the end of the bone do not undergo hypertrophy but remain as permanent cartilage, giving rise to the articular cartilage. In OA, however, the articular cartilage aberrantly undergoes hypertrophy and subsequent endochondral ossification, resulting in joint degeneration.2
There is no disease-modifying OA drug (DMOAD) currently available to patients that can halt disease progression and reverse the damage caused by OA.6 ,7 Commonly prescribed OA medications include non-steroidal anti-inflammatory drugs, analgesics, locally administered corticosteroids and viscosupplementation, which provide only symptomatic relief and eventually result in the need for surgical intervention. To date, most attempts to develop DMOADs have focused on stopping or delaying the degeneration of articular cartilage with little success.1 Indeed, repair of degenerative articular cartilage may provide a better therapeutic solution for patients with OA in clinical applications rather than stopping OA progression. We have identified a new potential DMOAD candidate with strong chondrogenic activity and tested its efficacy as a DMOAD using an OA animal model and investigated its mechanism of action.
Materials and methods
OA model
The experimental OA model was performed on 8-week-old male mice, as previously described.8 Briefly, under general anaesthesia, the medial collateral ligament was transected and the medial meniscus removed using a surgical microscope. A sham operation was performed on the contralateral knee joint using the same approach with no ligament transection or meniscectomy. Each of the prevention and repair models had two groups: (1) TD-198946-treated animals and (2) saline-treated animals (n=8 mice in each group). In all the mice tested the left knee joints underwent the operation and the right knee joints were sham-operated. For intra-articular injection, TD-198946 was dissolved at 100 nM in a solution of 45% 2-hydropropyl-b-cyclodextrin (HBC, Sigma)9 in saline (sterile 0.9% NaCl; Otsuka Pharmaceutical Factory). Mice were re-anaesthetised and given a 10 µl intra-articular injection of TD-198946 or saline immediately after surgery (prevention model) or 4 weeks following surgery (repair model) every 5 days for 8 or 4 weeks, respectively.
Histological analyses
Eight weeks after surgery the mice were killed and OA severity was quantified by the OARSI histopathology grading system (score 0–24)10 by two blinded independent observers. Sections were stained with safranin O-fast green or methylene blue using standard protocols. For immunohistochemistry, sections were incubated with antibodies to Runx1 (H-65; Santa Cruz Biotechnology), Sox9 (H-90; Santa Cruz Biotechnology), Sox6 (H-95; Santa Cruz Biotechnology), Sox5 (H-90; Santa Cruz Biotechnology), Col10a1 (LSL), Col1a1 (Santa Cruz Biotechnology) and Mmp-13 (Chemicon) diluted 1 : 500 in blocking reagent and detected with a CSAII Biotin-free Tyramide Signal Amplication System (Dako). For Col10a1, sections were treated with hyaluronidase (25 mg/ml in phosphate buffer saline) for 30 min. Sections were counterstained with methylgreen.
Other analyses
Construction of expression vectors, cell culture, screening of small compound libraries, real-time (RT)-PCR analysis, biochemical measurement of glycosaminoglycans, metatarsal organ culture, staining, microarray analysis, luciferase reporter assay, electrophoretic mobility shift assay, chromatin immunoprecipitation (ChIP) assay and animals and materials are described in the online supplementary materials and methods.
Statistical analysis
Data are reported as the mean±SEM of at least three independent experiments performed in triplicate. We performed statistical analyses of experimental data with the unpaired two-tailed Student t test. p Values <0.05 were considered significant.
Results
Identification of TD-198946 as a strong inducer of chondrogenic differentiation
We screened 2500 natural (InterBioScreen, Chernogolovka, Russia) and synthetic small compounds (Takeda Chemical Industries, Osaka, Japan) as potential chondrogenic agents via four steps using the Col2GFP-ATDC5 system (see online supplementary figure S1)11 and identified a thienoindazole derivative (TD-198946) that could potently increase toluidine blue staining after 7 days (figure 1A). A luciferase assay revealed that TD-198946 induced stronger COL2A1 promoter activity than insulin in ATDC5 cells (see online supplementary figure S2A). In C3H10T1/2 cells, ATDC5 cells and primary mouse chondrocytes, TD-198946 (10−10–10−5 M) dose-dependently stimulated endogenous expression of the chondrocyte markers Col2a1 and Acan, with maximum effects around 10−7–10−6 M, greater than the effects seen in combination with Sox5, Sox6 and Sox9 overexpression (dotted lines, figure 1B). Furthermore, TD-198946 enhanced cartilage matrix synthesis (alcian and toluidine blue stainings) in cultured C3H10T1/2 cells in both a dose- and time-dependent manner, and GAG content (figure 1C) without affecting cell proliferation (see online supplementary figure S2B).

Identification of TD-198946 as a potent chondrogenic agent. (A) Left: Green fluorescent protein (GFP) fluorescence of Col2GFP-ATDC5 cells after 2 and 7 days of culture with insulin (10 μg/ml), TD-198946 (10−6 M) or vehicle. BF, bright field. TB, toluidine blue staining. Scale bar 200 μm. Right: Chemical structure of TD-198946. (B) Dose-dependent effects of TD-198946 on mRNA of Col2a1 and Acan in C3H10T1/2 cells, ATDC5 cells and primary mouse chondrocytes for 7 days. Adenoviral overexpression in combination with Sox5, Sox6 and Sox9 are compared (dotted lines). (C) Alcian blue and toluidine blue stainings (left) and quantitatively analysed GAG content (right) in C3H10T1/2 cells for 7 and 14 days of culture with TD-198946 (10−12, 10−9 and 10−6 M) or the control vehicle (V). *p<0.01 vs vehicle. (D) Alcian blue, toluidine blue and alizarin red stainings and mRNA of Col2a1, Col10a1, Col1a1, Alp and Mmp13 in ATDC5 cells cultured with TD-198946 (10–7 M), insulin (10 μg/ml) or vehicle for 21 days. (E) In situ hybridisation of Col10a1 and von Kossa staining of metatarsal limbs derived from mouse embryos (E15.5) cultured with TD-198946 (10–7 M) or vehicle for 7 days. Red, blue and green bars indicate proliferative, hypertrophic and ossified zones, respectively; the percentages of these zones over the total length of the limbs are shown (graph). (F) Alcian blue and alizarin red stainings and mRNA of Col2a1, Col10a1 and Col1a1 in mouse dedifferentiated chondrocytes (P3) cultured with TD-198946 (10−7 M), bone morphogenetic protein-2 (BMP2, 100 ng/ml), transforming growth factor-β1 (TGF-β1, 1 ng/ml) or vehicle for 7 and 14 days. *p<0.01 vs vehicle. Data are expressed as mean±SD (error bars) for 4 wells/group.
Inhibitory effects of TD-198946 on hypertrophy and dedifferentiation
We next examined the effect of TD-198946 on hypertrophy and dedifferentiation of chondrocytes. In ATDC5 cells, TD-198946 enhanced chondrogenic differentiation (alcian blue, toluidine blue stainings and Col2a1 expression) without affecting hypertrophy (alizarin red staining, Col10a1, Alp and Mmp13 expressions) or dedifferentiation (Col1a1 expression) (figure 1D). Contrarily, insulin enhanced all three sets of markers. These effects were confirmed in an organ culture of metatarsal limbs derived from mouse embryos (figure 1E) where TD-198946 delayed the start of the hypertrophic zone. The hypertrophic zone was compressed with decreased mineralisation while the proliferative zone was expanded.
The effect of TD-198946 was then compared with other widely used agents for cartilage regeneration—namely, bone morphogenetic protein-2 (BMP2) and transforming growth factor-β1 (TGF-β1). Using dedifferentiated primary mouse chondrocytes (figure 1F), TD-198946 strongly induced chondrogenic redifferentiation (alcian blue staining and Col2a1 expression) throughout 14 days of culture without affecting hypertrophy or dedifferentiation (alizarin red staining, Col10a1 or Col1a1 expressions). By comparison, BMP2 showed temporal chondrogenic activity at 7 days and enhanced hypertrophy or dedifferentiation at 14 days, while TGF-β1 had no positive effect on chondrogenesis. Overall, TD-198946 is a stronger chondrogenic agent than BMP2 and TGF-β1 without causing hypertrophy and dedifferentiation. We further confirmed that TD-198946 is not involved in the activity of BMP,12 TGF-β13 or canonical Wnt signals (see online supplementary figure S3).14
Prevention and repair of induced OA by intra-articular injection of TD-198946
We next examined the effects of TD-198946 in a surgically-induced OA mouse model. Periodic injections of TD-198946 or saline were administered into the knee joints immediately (prevention model) or 4 weeks after surgery (repair model) for up to 8 weeks. Representative histological sections and OARSI histopathology grading revealed that TD-198946-treated joints showed fewer osteoarthritic changes than untreated joints (figure 2A,B). In addition, TD-198946 had no influence on the normal mouse knee joint (see online supplementary figure S4).

Effects of TD-198946 treatment on surgically-induced osteoarthritis (OA). (A) Safranin-O stained sections of mouse medial tibial cartilage obtained from prevention and repair models. (B) Quantification of OA on femoral and tibial cartilage by OARSI histopathology grading. Data are expressed as mean±SEM (n=8). *p<0.05 vs saline. Scale bar 100 μm.
Identification of Runx1 as a molecular target of TD-198946
The chondrogenic effects of TD-198946 were next assessed using microarray by expression profiling on mRNA isolated from C3H10T1/2 cells treated with TD-198946 or vehicle (data sets S1 and S2 in online supplement). The profiling revealed 196 genes with more than 10-fold upregulation by TD-198946 (dataset S1; see online supplementary figure S5A). Using gene ontology (GO) analysis of a set of the 196 genes (dataset S2), the most over-represented term was ‘multicellular organismal development’ (GO:0007275; p=2.45×10−5) which was associated with 59 genes, suggesting that TD-198946 exerted development- or differentiation-related effects (see online supplementary figure S5B). Runx1 ranked as the top of six transcription-related genes associated with the GO term (see online supplementary figure S5C). Sox5 and Sox6 ranked as second and third, both of which have important roles as a co-factor of Sox9 during chondrogenesis.
Induction of chondrogenic differentiation by Runx1
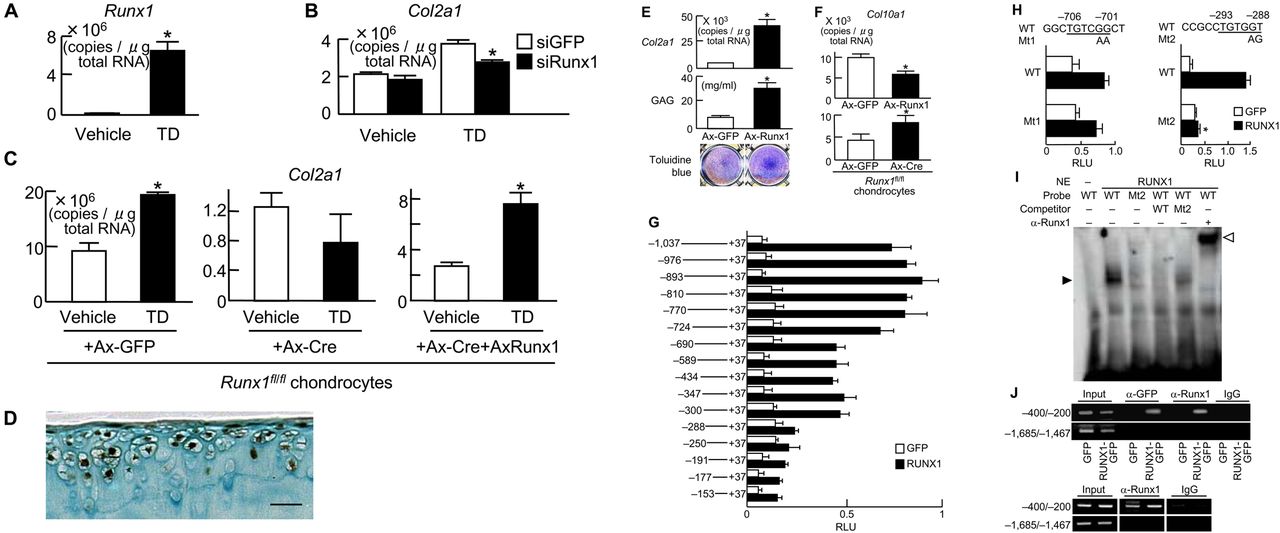
Induction of Runx1 mRNA by TD-198946 was identified in C3H10T1/2 cells (figure 3A). Small interfering RNA (siRNA) of Runx1 confirmed the role of Runx1 in mediating the chondrogenic effects of TD-198946 with significant inhibition of TD-198946-induced Col2a1 in C3H10T1/2 cells (figure 3B). Furthermore, in primary chondrocytes derived from Runx1fl/fl mice,15 Cre-mediated deletion of Runx1 cancelled the TD-198946-stimulated expression of Col2a1, which could be restored by the adenoviral overexpression of Runx1 (figure 3C).

Runx1 as a target of TD-198946. (A) Runx1 mRNA levels in C3H10T1/2 cells cultured with TD-198946 (10−7 M) or vehicle for 7 days. *p<0.01 vs vehicle. (B) Col2a1 mRNA levels in C3H10T1/2 cells cultured with TD-198946 (10−7 M) or vehicle for 2 days after Runx1 siRNA transfection or the control green fluorescent protein (GFP). *p<0.01 vs siGFP. (C) Col2a1 mRNA levels in primary mouse chondrocytes from Runx1-floxed (Runx1fl/fl) mice cultured with TD-198946 (10−7 M) or vehicle for 2 days after adenoviral (Ax) infection with GFP, Cre, or Cre+Runx1; *p<0.01 vs vehicle. (D) Runx1 protein in normal knee cartilage from wild-type 16-week-old mice. Scale bars 100 μm. (E) Col2a1 mRNA, GAG content and toluidine blue staining in cultured C3H10T1/2 cells adenovirally (Ax)-infected with Runx1 or control GFP. *p<0.01 vs Ax-GFP. (F) Col10a1 mRNA in chondrocytes from Runx1-floxed (Runx1fl/fl) mice in 3-D alginate bead cultures after adenoviral (Ax) infection with GFP, Runx1 or Cre. *p<0.01 vs Ax-GFP. (G) Deletion analysis using luciferase-reporter constructs containing the 5′-flanking sequence from −1031 to +37 of the COL2A1 gene and deletion fragments in HuH-7 cells transfected with RUNX1 or control GFP; *p<0.01 vs GFP. (H) Site-directed mutagenesis (Mt1 and Mt2) using luciferase-reporter constructs containing the Runx motifs in −724/−690 (left) and −300/−288 (right) of the COL2A1 gene in HuH-7 cells. (I) Electrophoretic mobility shift assay for specific binding (closed arrowhead) of wild-type (WT) oligonucleotide probe (–304/–275) containing Runx motif above or mutated probe (Mt2) with nuclear extract (NE) of COS-7 cells overexpressing Runx1. Cold competition with 50-fold excess of unlabelled WT or Mt2 probes and the supershift by α-Runx1 (open arrowhead). (J) Chromatin immunoprecipitation assay on HeLa cells transfected with GFP or RUNX1-GFP (top) and primary mouse chondrocytes (bottom). Cell lysates were amplified by primer sets spanning the Runx motif (−400/−200) or not (−1685/−1467) before (input) and after immunoprecipitation with α-GFP or α-Runx1 antibodies or non-immune IgG. Data are expressed as mean (bars)±SD (error bars) for 4 wells/group.
In normal mouse knee cartilage, Runx1 was strongly expressed in the superficial and middle zones of cartilage above the tidemark but not in the mineralised cartilage (figure 3D). In cultured C3H10T1/2 cells, Runx1 overexpression potently enhanced Col2a1 expression, GAG content and toluidine blue staining (figure 3E) but inhibited Col10a1 expression, which could be restored in primary chondrocytes derived from Runx1fl/fl mice by Cre-mediated Runx1 knockdown (figure 3F).15 These data indicate that Runx1 is an inducer of chondrogenic differentiation and a suppressor of the subsequent hypertrophy, which is true of the effect of TD-198946.
We further examined the mechanism underlying the transactivation of COL2A1 by Runx1. Deletion analyses of the COL2A1 promoter (−1031/+37 used in the Col2GFP-ATDC5 system) predicted the responsive elements to be located between −724 and −690 bp, and between −300 and −288 bp, both of which contain Runx motifs (figure 3G).16 Site-directed mutagenesis in the downstream Runx motif, but not in the upstream motif, caused significant suppression, indicating that the −293/−288 site is the core responsive element of Runx1 (figure 3H). Electrophoretic mobility shift assay (EMSA) revealed the binding of nuclear extracts of Runx1-overexpressed COS-7 cells with the −293/−288 oligonucleotide probe (figure 3I). The binding specificity was verified by cold competition with excess amounts of the unlabelled wild-type probe, but not with that of the mutated probe, as well as by the supershift with an antibody to Runx1. In vivo binding of exogenous and endogenous Runx1 to this Runx motif was confirmed using ChIP assay in Runx1-transfected HeLa cells and primary mouse chondrocytes (figure 3J), respectively.
To investigate the in vivo role of Runx1 in OA, we examined Runx1 expression in articular cartilage obtained from surgically-induced mouse OA prevention and repair models treated with TD-198946. Immunohistochemistry showed substantial expression of Runx1 in TD-198946-treated cartilage but not in saline-treated cartilage (figure 4A,B and online supplementary figure S6), similar to those of Sox9, Sox6 and Sox5. Sox9, Sox6 and Sox5 expressions were not detected in the control. Col10a1, Col1a1 and Mmp-13 expressions were not detected in TD-198946-treated cartilage but upregulated in the control. Finally, to assess the role of Runx1 in human OA, we investigated the expression of Runx1, Sox6, Sox9 and Col10a1 in OA and normal cartilage from a patient. Runx1, Sox6, Sox9 and Sox5 were downregulated in OA cartilage whereas Col10a1 was upregulated compared with normal cartilage (figure 4C and online supplementary figure S7). We also confirmed that TD-198946 treatment induced redifferentiation of dedifferentiated human normal chondrocytes and OA-like chondrocytes in vitro (see online supplementary figures S8 and S9).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effects of TD-198946 treatment on Runx1, Sox6, Sox9 and Col10a1 expression in osteoarthritis (OA). (A) Methylene blue-stained sections of the mouse medial tibial cartilage obtained from prevention and repair models. Scale bar 100 μm. (B) Immunostainings of Runx1, Sox6, Sox9 and Col10a1 from the inset box of (A). Scale bar 50 μm. (C) Safranin-O staining and immunostaining of Runx1, Sox6, Sox9 and Col10a1 in normal and OA regions of tibial cartilage obtained from a patient who underwent total knee arthroplasty. Scale bar 100 μm.
Discussion
In this study we identified a novel DMOAD candidate, TD-198946, which strongly induced chondrogenic differentiation without promoting hypertrophy. When administered directly into the joint space, the compound successfully prevented and repaired the degeneration of articular cartilage in a surgically-induced mouse model of OA. The compound exerted its effect by regulating Runx1 expression, an inducer of chondrogenic differentiation and a suppressor of the subsequent hypertrophy, which possibly underlies the therapeutic effect of TD-198946 on OA.
Several recent reports on new candidate DMOADs have emerged. However, unlike TD-198946, the major action of these agents is to block the activation of the degradation enzyme and/or to inhibit aberrant hypertrophy of articular cartilage by targeting signalling molecules involved in articular cartilage catabolism. In a surgically-induced model of OA, intra-articular injections of antibodies specific for syndecan-4, which is involved in aggrecan cleavage by ADAMTS-5 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif 5), prevented cartilage destruction.17 Similarly, intraperitoneal injections of an inhibitor of Hedgehog (Hh) signalling, which is activated in OA, reduced the severity of OA by inhibiting cartilage destruction, potentially through Runx2 regulation, which in turn regulates ADAMTS-5.18 Furthermore, the subcutaneous injection of recombinant human parathyroid hormone (1–34) (teriparatide) immediately or 8 weeks after OA induction partially reduced the severity of OA by suppressing inappropriate articular cartilage hypertrophy.19 Moreover, during the review process of our paper, another small molecule, kartogenin, was reported to induce chondrogenesis by regulating the CBFβ-RUNX1 transcriptional programme.20 Runx1 may be a candidate molecule for the treatment of OA.
The major action of TD-198946 is distinct from the actions of the above strategies: it strongly induces chondrogenic differentiation of progenitors, as evidenced by a marked increase in the chondrogenic markers type II collagen and aggrecan, and it does this without promoting hypertrophy, which is often and undesirably observed with conventional chondrogenic agents including BMPs,21 TGF-β,13 insulin21 and insulin-like growth factor-1.22 TD-198946 may help rejuvenate articular cartilage by shifting the differentiation balance from late differentiation (hypertrophy) towards early differentiation (chondrogenesis). TD-198946 is the first small molecule compound to have both reparative and preventive effects in a mouse degenerative articular cartilage model. In the clinic, OA is usually diagnosed when the disease has substantially progressed and, because articular cartilage has a poor regenerative capacity, stopping or delaying cartilage degeneration alone is unlikely to treat the disorder sufficiently.
The target molecule of TD-198946 is distinct; it strongly induced Runx1, which is known to be a chondrogenic factor.5 ,15 In both mouse and human, Runx1 is expressed in adult articular cartilage and its expression is downregulated in OA cartilage. In our study, Runx1 expression was restored by TD-198946 treatment, and our gain and loss of function experiments revealed that Runx1 mediated the chondrogenic action of TD-198946. Runx1 directly enhanced the promoter activity of COL2A1 through specific binding to a Runx motif. In addition, our study indicates that Runx1 suppresses hypertrophy. On the other hand, Runx2 and Runx3 are essential for hypertrophy and maturation of chondrocytes.23 We have previously reported that Runx1 induces Sox5, Sox6 and Sox9 expression during chondrogenesis.15 TD-198946 seems to induce Sox5 and Sox6 indirectly via Runx1, judging from the data using chondrocytes from Runx1 knock-out mice (see online supplementary figure S10). These transcription factors, which stimulate chondrogenesis and suppress hypertrophy,24 were also induced by TD-198946, albeit to a lesser extent. Our findings indicate that, in addition to its direct effect on chondrogenesis, Runx1 activity may also be mediated by the Sox transcription factors.
Together, our data suggest that TD-198946 is a novel DMOAD candidate. Given its unique actions and target molecule, it can be used alone or synergistically with other agents. We also show that Runx1 is a promising target for the treatment and development of other DMOADs.
Acknowledgments
We thank A Kimura and S Takeda for providing Runx1fl/fl mice.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
- Data supplement 3 - Online supplement
- Data supplement 4 - Online supplement
- Data supplement 5 - Online supplement
- Data supplement 6 - Online figures
- Data supplement 7 - Online legends
Footnotes
-
Funding This study was supported by a grant-in-aid for scientific research from the Japanese Ministry of Education, Culture, Sports, Science and Technology (#21659460 and #22689052), the Programme for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO, Project ID 06-34), the University of Tokyo Center for NanoBio Integration (CNBI programme) and the Center for Medical System Innovation (CMSI programme), the Japan Society for the Promotion of Science (JSPS) through the ‘Funding Programme for World-Leading Innovative R&D on Science and Technology (FIRST Programme)’ initiated by the Council for Science and Technology Policy (CSTP). The sponsors had no role in study design, data collection, data analysis, data interpretation or writing of the manuscript.
-
Contributions FY, UC and HK conducted the project planning. FY, AF, YH and HH performed the experiments. FY, TI, TS, HC, MH, SO, TT, UC and HK handled data analysis. FY, SO, HK and UC wrote the manuscript.
-
Comepeting interests None.
-
Patient consent Obtained.
-
Ethics approval Knee cartilage samples were obtained from OA patients undergoing total knee arthroplasty.
-
Provenance and peer review Not commissioned; externally peer reviewed.