Article Text

Abstract
Objectives To assess long-term golimumab efficacy/safety in patients with active psoriatic arthritis (PsA).
Methods Adult PsA patients (≥3 swollen, ≥3 tender joints, active psoriasis) were randomly assigned to subcutaneous injections of placebo, golimumab 50 mg or 100 mg every 4 weeks (q4wks) through week 20. All patients received golimumab 50 or 100 mg beginning week 24. Findings through 2 years are reported. Efficacy evaluations included ≥20% improvement in American College of Rheumatology (ACR20) response, good/moderate response in Disease Activity Scores incorporating 28 joints and C-reactive protein (DAS28-CRP), ≥75% improvement in Psoriasis Area and Severity Index (PASI75) and changes in PsA-modified Sharp/van der Heijde scores (SHS).
Results Golimumab treatment through 2 years was effective in maintaining clinical response (response rates: ACR20 63%–70%, DAS28-CRP 77%–86%, PASI75 56%–72%) and inhibiting radiographic progression (mean change in PsA-modified SHS in golimumab-treated patients: −0.36), with no clear difference between doses. No new safety signals were identified through 2 years. With the study's tuberculosis screening and prophylactic measures, no patient developed active tuberculosis through 2 years.
Conclusions Golimumab 50 and 100 mg for up to 2 years yielded sustained clinical and radiographic efficacy when administered to patients with active PsA. Increasing the golimumab dose from 50 to 100 mg q4wks added limited benefit. Golimumab safety through up to 2 years was consistent with other antitumour necrosis factor α agents used to treat PsA. Treatment of patients with latent tuberculosis identified at baseline appeared to be effective in inhibiting the development of active tuberculosis.
- Psoriatic Arthritis
- Anti-TNF
- Spondyloarthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA), a manifestation of psoriatic disease, involves inflammation of the peripheral/axial joints, entheses and usually concomitant skin/nail psoriasis. Antitumour necrosis factor (anti-TNF) α agents are effective PsA treatments.1 Despite the abundance of long-term efficacy/safety data for anti-TNF agents in rheumatoid arthritis (RA), corresponding long-term data in PsA are relatively sparse. Also, with the exception of the current golimumab trial, no other trial has evaluated, in a randomised fashion, two different dosing regimens of the same anti-TNF agent in active PsA.
Golimumab is a human anti-TNF monoclonal antibody2 administered subcutaneously every 4 weeks (q4wks) that has demonstrated efficacy in RA2–7 and ankylosing spondylitis.8 We previously detailed golimumab efficacy/safety through weeks 24 and 52 of the GO-REVEAL phase III, randomised, double-blind, placebo-controlled trial in 405 patients with active PsA.9 ,10 In GO-REVEAL, subcutaneous golimumab (50 or 100 mg q4 wks) significantly improved arthritic manifestations of PsA and associated skin disease,9 and reduced radiographic progression10 versus placebo through week 24. No dose differentiation was observed, with the exception of numerically greater responses in skin-related endpoints with the higher golimumab dose.9 Beginning at week 24, all patients received golimumab 50 or 100 mg. Patients then entered the open-label long-term study extension after week 52, in which the golimumab dose could be increased from 50 to 100 mg q4ws. Efficacy/safety data of long-term PsA treatment with two different golimumab doses through 2 years are reported herein.
Patients and methods
Patients
Patients were naive to anti-TNF therapy, had active PsA (≥3 swollen, ≥3 tender joints) and had plaque psoriasis (qualifying lesion ≥2 cm in diameter) despite therapy with disease-modifying antirheumatic drugs (DMARDs) or non-steroidal anti-inflammatory drugs (NSAIDs). Stable doses of methotrexate (MTX), NSAIDs and corticosteroids (prednisone ≤10 mg/day) were allowed through week 52, after which non-biologic DMARD/immunosuppressive, corticosteroid, NSAID and topical therapies could be adjusted. Light therapy was prohibited throughout the study. Patients with latent tuberculosis identified at screening via purified protein derivative skin or whole blood interferon-γ-based QuantiFERON-TB Gold testing (Cellestis, Inc, Valencia, California, USA) could participate if treated prior to/concurrent with study treatment. Institutional review board or ethics committee approval and patient written informed consent were obtained prior to study procedures.
Study design
Overall, 405 patients were randomised (1:1.3:1.3) to receive blinded subcutaneous injections of placebo, golimumab 50 mg or golimumab 100 mg at weeks 0, 4, 8, 12, 16 and 20, with stratification by baseline MTX use. Golimumab (Janssen Research & Development, LLC, Spring House, Pennsylvania, USA) and placebo were supplied as sterile liquid for injection. At week 16, patients with <10% improvement in both swollen and tender joint counts could early escape from placebo → golimumab 50 mg or golimumab 50 → 100 mg; patients randomised to golimumab 100 mg continued blinded 100 mg. Starting at week 24, all patients still receiving placebo crossed over to golimumab 50 mg. Thus, all study participants received blinded golimumab 50 or 100 mg q4wks between week-24 and week-52 database locks. Subsequently, patients received open-label subcutaneous golimumab (50 or 100 mg) injections q4wks and those receiving 50 mg could escalate the dose to 100 mg q4wks based on investigator judgment. Patients receiving golimumab 100 mg had no further dose escalation. Patients discontinuing golimumab treatment were followed for approximately 4 months and contributed follow-up data to efficacy/safety analyses. Week 104 was the last evaluation for this report.
Study endpoints
The study's co-primary endpoints, the proportion of patients achieving ≥ 20% improvement in American College of Rheumatology (ACR20) response criteria11 at week 149 and change from baseline in total PsA-modified Sharp/van der Heijde score (SHS) at week 24,10 have been reported. ACR responses and Disease Activity Scores incorporating 28 joints and C reactive protein (DAS28-CRP)12 ,13 were assessed through week 104. Centrally digitised radiographic images obtained at weeks 0, 52 and 104 were scored by two independent readers in random order without knowledge of time point, patient identity or treatment using a PsA-modified SHS.14 ,15
The extent/severity of psoriasis were assessed by Psoriasis Area and Severity Index (PASI) scores (range 0–72)16 among patients with ≥ 3% of baseline body surface area (BSA) psoriasis skin involvement. Physical function was assessed using the disability index of the Health Assessment Questionnaire (HAQ).17 ,18 Dactylitis was scored in each of 20 hand/foot digits from 0 to 3 (0=no dactylitis; 3=severe dactylitis; range 0–60). Entheses tenderness was scored for 15 sites (0=absent; 1=present; range 0–15) using the PsA-modified (left and right insertion of plantar fascia added) Maastricht Ankylosing Spondylitis Enthesitis Score.19 Nail psoriasis was assessed with the Nail Psoriasis Severity Index (NAPSI)20 for a target fingernail representing the worst baseline fingernail psoriasis. The target nail was graded for nail matrix psoriasis and nail bed psoriasis from 0 to 4 (0=no nail psoriasis; 4=nail psoriasis in four quadrants; range 0–8). For enthesitis, dactylitis and fingernail psoriasis, a score ≥ 1 denoted ‘presence’.
Safety evaluations included adverse events (AEs) and routine laboratory analyses. Patients were questioned regularly about symptoms suggesting active TB. Antibodies to golimumab3 and trough golimumab concentrations were determined from serum samples collected prior to injections at selected visits.
Statistical analyses
Sample size estimation has been detailed.9 Due to protocol-allowed changes in the golimumab regimen throughout the study and to account for patient dropout, clinical efficacy data were analysed using two approaches detailed below. Descriptive statistics were employed to summarise all findings.
-
Intent-to-treat (ITT) analysis by randomised group, irrespective of treatment changes during the study. Missing data for ACR, DAS28, PASI, dactylitis, enthesitis, NAPSI and HAQ were imputed using last-observation-carried-forward methodology; patients who discontinued treatment due to lack of efficacy were considered non-responders for ACR/DAS28 assessments. For radiographic data, modified ITT analyses included patients with baseline and ≥ 1 postweek 52 SHS available. Missing week 104 PsA-modified SHSs were imputed using last-observation-carried-forward methodology between weeks 52 and 104. Intraclass correlation coefficients were determined to assess correlation/reliability between radiograph readers.
-
Analyses by treatment received for patients who increased the golimumab dose from 50 → 100 mg after the week-52 database lock (per investigator judgment) and who had ≥12 weeks of follow-up.
Safety summaries included all treated patients, and individual AEs were attributed to treatment received at event onset. The incidences of death, serious infection, malignancy and major adverse cardiovascular events (MACE; defined as cardiovascular deaths or cardio/cerebrovascular serious AEs) were calculated per 100 patient-years (/100 pt-yrs) to account for differences in length of follow-up between treatment groups. Observed incidences of malignancy were compared with those expected in the general US population per the Surveillance, Epidemiology and End Results (SEER) database.21 Standardised incidence ratios were calculated as ‘observed’ (GO-REVEAL) divided by ‘expected’ (SEER database) cases of malignancy, and exact 95% CIs were determined. Non-melanoma skin cancer (NMSC) is not included in the SEER database and thus not included in the comparative analyses.
Results
Patient disposition and baseline characteristics
Among 555 adults screened at 58 sites,9 405 were randomised and treated. Consent was obtained for the first patient on 12 December 2005; the last patient completed week 104 on 18 November 2008.
Patient disposition through week 249 and week 5210 have been reported. Among 405 randomised patients, 335 (83%) continued study treatment through week 104. The most common reasons for study agent discontinuation were AEs (6%–8% across randomised groups) and unsatisfactory therapeutic response (3%–5%) (table 1). Radiographic images and corresponding scores were available for 397 (98%), 361 (89%) and 331 (82%) patients at weeks 0, 52 and 104, respectively.
Patient disposition through week 104
Baseline patient/disease characteristics were generally consistent among randomised treatment groups (table 2), with the exception of higher baseline SHS in patients receiving MTX at baseline. Despite the investigator's ability to adjust concomitant non-biologic DMARD/immunosuppressive, corticosteroid and/or NSAID treatment starting at week 52, the proportions of patients taking these medications and the mean doses at week 104 were generally similar or slightly lower versus baseline (tables 2 and 3).
Baseline patient characteristics and concomitant medication use
Summary of efficacy and concomitant medication use at week 104 by randomised treatment group
Efficacy results based on ITT analyses (see ‘Statistical analyses’)
ACR and DAS28-CRP responses
Among randomised patients, week 104 response rates were 63%–70%, 46%–51% and 29%–36% for ACR20, ACR50 and ACR70, respectively (figure 1A). ACR responses were similar irrespective of baseline MTX use (figure 1B,C). Also, 77%–86% of randomised patients achieved DAS28-CRP responses of good/moderate; mean DAS28-CRP scores at week 104 were 2.8–3.1 (table 3).
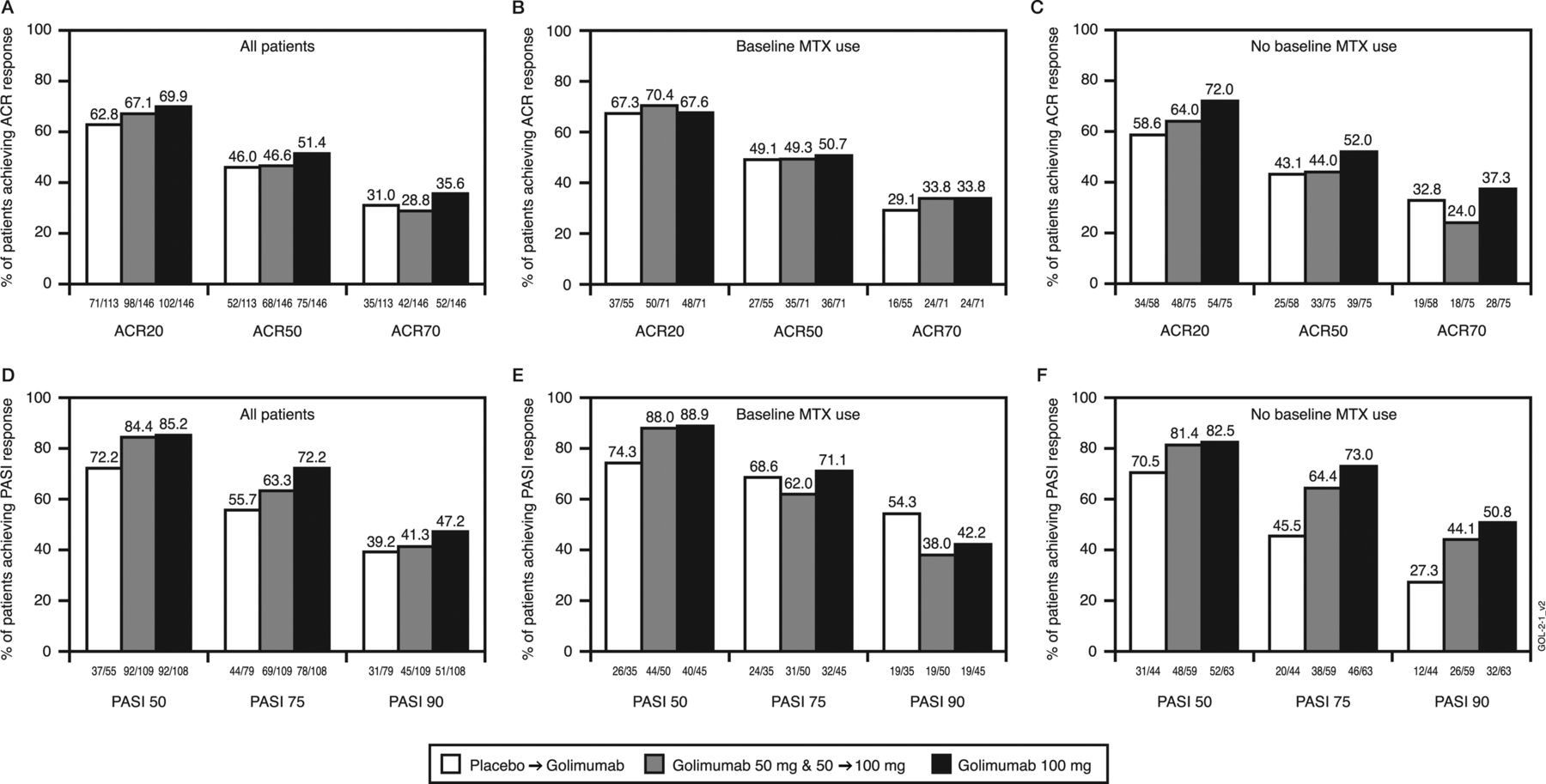
The proportions of patients achieving clinical improvement, defined by at least 20%, 50% and/or 70% improvement in the American College of Rheumatology (ACR20, ACR50, and ACR70, respectively) response criteria ((A) all patients, (B) patients with methotrexate (MTX) use at baseline, (C) patients with no MTX use at baseline) or at least 50%, 75% and/or 90% improvement in the Psoriasis Area and Severity Index (PASI50, PASI75 and PASI90, respectively) response criteria among randomised patients with baseline psoriasis involving ≥3% body surface area ((D) all patients, (E) patients with MTX use at baseline, (F) patients with no MTX use at baseline). Patients were assessed according to randomised treatment group using an intent-to-treat analysis and missing data imputation rules were applied. Placebo patients include all patients randomised to the placebo arm, including those who early escaped at week 16 or crossed over at week 24 to receive golimumab 50 mg or dose escalated after the week-52 database lock to receive golimumab 100 mg. Patients in the golimumab 50-mg group include all patients who were randomised to the golimumab 50-mg arm including those patients who early escaped at week 16 or dose escalated after the week-52 database lock to receive golimumab 100 mg. Patients in the golimumab 100-mg group include all patients randomised to the golimumab 100-mg arm; these patients had no change in study treatment through week 104.
Dactylitis and enthesitis
Through week 104, mean per cent improvements in dactylitis score were 67%–85% in patients with baseline dactylitis and in the PsA-modified Maastricht Ankylosing Spondylitis Enthesitis Score were 40%–60% among patients with baseline enthesitis (table 3).
Skin and nail responses
PASI75 response (figure 1D) was achieved at week 104 in 56%–72% of randomised patients with baseline psoriasis involving ≥3% BSA. PASI responses were similar irrespective of baseline MTX use (figure 1E,F). Mean per cent improvements in NAPSI scores ranged from 61% to 70% among patients with baseline fingernail psoriasis (table 3).
Physical function
Clinically meaningful improvements in physical function (HAQ decrease ≥ 0.3)22 were observed at week 104 for 53%–59% of randomised patients. Mean week 104 HAQ scores were 0.6–0.7 (table 3).
Radiographic response
The two readers demonstrated good agreement in radiographic image scoring, with intraclass correlation coefficients of 0.96 at baseline, week 52 and week 104. At week 104, mean±SD changes from baseline in PsA-modified SHS were 0.08±3.19, –0.39±2.04 and −0.32±1.87 in the placebo, 50 mg and 100 mg groups, respectively (figure 2A). Patients receiving MTX at baseline demonstrated numerically less progression at week 104 than patients not receiving MTX (table 3, figure 2). Mean changes in SHS erosion and joint-space-narrowing subscores are presented in table 3.

{kind=link}
{kind=link}
Psoriatic arthritis (PsA)-modified Sharp/van der Heijde score (SHS). (A–C) Changes from baseline in the total PsA-modified SHS at week 104 by randomised treatment group and baseline methotrexate (MTX) use. (A) All patients, (B) patients receiving MTX at baseline and (C) patients not receiving MTX at baseline. Figures include patients who had a total modified SHS at baseline and after week 52. Means are represented by solid lines, medians by dotted lines and IQRs by bars. Missing data rules were applied. (D–F) Empirical cumulative distribution function of change from baseline in total PsA-modified SHS at week 104 by randomised group and baseline MTX use. (D) All patients, (E) patients receiving MTX at baseline and (F) patients not receiving MTX at baseline. The cumulative distribution function plots include patients who had scores at baseline and after week 52. No missing data imputation was applied (ie, plots exclude two, three and three patients in the placebo, golimumab 50-mg and golimumab 100-mg groups, respectively, who were included in the analysis of change from baseline to week 104 in figure 2A–C). All panels: placebo patients include all patients randomised to the placebo arm, including those who early escaped at week 16 or crossed over at week 24 to receive golimumab 50 mg or dose escalated after the week-52 database lock to receive golimumab 100 mg. Patients in the golimumab 50-mg group include all patients who were randomised to the golimumab 50-mg arm including those who early escaped at week 16 or dose escalated after the week-52 database lock to receive golimumab 100 mg. Patients in the golimumab 100-mg group include all patients randomised to the golimumab 100-mg arm; these patients had no change in study treatment through week 104.
Efficacy results for patients who received a golimumab dose increase (50 → 100 mg) (see ‘Statistical analyses’)
In all, 33 patients receiving golimumab 50 mg q4wks had their dose escalated based on the investigator's judgment to 100 mg q4wks after the week-52 database lock and had ≥12 weeks of follow-up postescalation. These patients generally had low disease activity prior to dose escalation, with a median (mean) DAS28-CRP score of 3.4 (3.3) and PASI score of 1.5 (3.2). Dose escalation provided limited benefit at 12 weeks following initiation of the higher golimumab dose, with more improvement observed in skin disease than arthritis (table 3).
Golimumab pharmacokinetics and antibodies to golimumab
Dose-proportional pharmacokinetics was generally observed through 2 years of treatment. Median serum golimumab concentrations were maintained over time and corresponded with the observed sustained clinical efficacy. Consistent with findings through week 249 and week 52,10 few patients developed antibodies to golimumab through week 104 (5.4% (21/388) among patients with ≥ 1 sample after the first administration of study agent). The development of antibodies to golimumab appeared to be less common in patients receiving (1.6% (3/191)) versus not receiving (9.1% (18/197)) baseline MTX and in patients who received golimumab 50 mg only (7/162, 4.3%) or 100 mg only (8/145, 5.5%) versus patients who increased the golimumab dose from 50 to 100 mg (6/81, 7.4%).
Safety results
AEs through week 104
Per protocol, no patient received placebo beyond week 24. The most commonly reported AEs in golimumab-treated patients were upper respiratory tract infection (23% of patients) and nasopharyngitis (16%). Infections occurred in 248 (63%) golimumab-treated patients. In general, no differences in the types of AEs were observed between golimumab 50 and 100 mg (table 4).
Summary of safety through week 104
Overall, 23 (6%) golimumab-treated patients discontinued study agent because of an AE, including 11 (4%) and 12 (5%) patients receiving golimumab 50 and 100 mg, respectively (table 4). AEs leading to discontinuation observed in > 1 patient overall included increased alanine aminotransferase (4–50 mg, 1–100 mg patients), increased aspartate aminotransferase (3–50 mg, 1–100 mg patients) and basal cell carcinoma (BCC, 1–50 mg, 3–100 mg patients).
Serious AEs were reported for 16 (6.5%) and 18 (8%) patients receiving golimumab 50 and 100 mg, respectively (table 4). Six patients had serious infections: two had an abscess and one had superficial thrombophlebitis while receiving golimumab 50 mg and one patient each had acute cholecystitis/sepsis, cellulitis and histoplasmosis while receiving golimumab 100 mg. The patient with histoplasmosis resided in an endemic area and was successfully treated with voriconazole. When assessed according to patient-years of follow-up, no increase in the incidence of serious infection was observed for golimumab- (0.84 (95% CI 0.17 to 2.45) for 50 mg; 1.20 (95% CI 0.33 to 3.07) for 100 mg) versus placebo- (9.41 (95% CI 2.56 to 24.08)) treated patients, although the analyses are limited by a relatively short duration of placebo treatment and small number of patients. No patient developed active tuberculosis through week 104, including 44 patients requiring tuberculosis prophylaxis following detection of latent disease at screening. Through week 104, one patient received treatment for latent tuberculosis newly diagnosed at week 36.
Four MACE cases were observed through week 104: acute myocardial infarction (100 mg), congestive cardiac failure (100 mg), myocardial infarction (50 mg) and myocardial ischaemia (placebo). The MACE incidence/100 pt-yrs indicated no increase with golimumab (0.28 (95% CI 0.01 to 1.56) for 50 mg; 0.60 (0.07 to 2.17) for 100 mg) versus placebo (2.35 (95% CI 0.06 to 13.10)) treatment.
Two golimumab 50 mg randomised patients died (one climbing accident, one small cell lung cancer) through week 52.10 No deaths occurred from weeks 52 to 104. When assessed by patient-years of follow-up, no increase in the incidence of death was observed for golimumab- (0.56 (95% CI 0.07 to 2.02) for 50 mg; 0.00 (95% CI 0.00 to 0.90) for 100 mg) versus placebo- (0.00 (95% CI 0.00 to 7.04)) treated patients.
Eight malignancies were documented through week 104, including colon cancer (placebo → golimumab 50 mg), prostate cancer (100 mg), two squamous cell lung cancers (1–50 mg, 1–100 mg) and four BCCs (1–50 mg, 3–100 mg). When assessed by patient-years of follow-up, the incidence of malignancies for golimumab-treated patients was numerically higher (see table 4) compared with patients receiving placebo (0.00/100 pt-yrs (95% CI 0.00 to 7.04)).
In an analysis comparing incidences of all malignancies (excluding NMSC) observed in GO-REVEAL and expected rates in the general US population, 95% CIs for the standardised incidence ratios contained 1 for all treatment groups (0.00 (95% CI 0.00 to 14.57) for placebo; 1.22 (95% CI 0.15 to 4.41) for 50 mg; 1.10 (95% CI 0.13 to 3.99) for 100 mg). Together, week 104 findings suggest no increase in malignancies among golimumab-treated patients, although analyses are limited by small sample size and short period of placebo follow-up.
Injection-site reactions, most commonly mild injection-site erythema, occurred in 31 (8%) patients through week 104 (table 4). No injection-site reaction was severe, serious or resulted in treatment discontinuation. No patient experienced anaphylactic or serum sickness-like reactions.
Discussion
We previously reported on the efficacy and safety of the human monoclonal anti-TNF agent golimumab, administered subcutaneously q4wks at doses of 50 or 100 mg, in patients with active PsA.9 ,10 Briefly, when compared with the control arm, golimumab-treated patients displayed significant and/or clinically meaningful improvements in all aspects of PsA, including peripheral arthritis, enthesitis, dactylitis and skin/nail psoriasis. In addition, consistent with other anti-TNF agents, golimumab treatment inhibited progression of radiographic damage in the hands and feet of patients with active PsA. The safety profile of golimumab was also consistent with other anti-TNF agents used to treat Patients with PsA.
Despite the lack of a control arm in the long-term study extension, findings through week 104 support the benefit of golimumab in maintaining clinical and radiographic responses observed in PsA. Patient attrition for all golimumab-treated patients was 17% through week 104, including 7% and 4% of patients discontinuing due to AEs and lack of efficacy, respectively. When analysed using ITT methodology with imputation of missing data, week 104 response rates were 67%–70% for ACR20, 47%–51% for ACR50 and 63%–72% for PASI75 (among patients with ≥3% of BSA psoriasis involvement). Similar clinical responses were observed among patients receiving and not receiving MTX at baseline.
Improvements were also observed in dactylitis, enthesitis and nails psoriasis, and golimumab-treated patients also continued to exhibit inhibition of structural damage, with a mean change in SHS from baseline to week 104 of −0.32 to −0.39 in patients randomised to golimumab. No clear differences between golimumab 50 and 100 mg were observed in long-term efficacy findings. In general, patients receiving MTX at baseline had smaller changes from baseline to week 104 in total SHS compared with patients not receiving MTX, despite having more radiographic damage at baseline. Among patients who crossed over from placebo → golimumab 50 mg, slightly fewer patients achieved some of the clinical responses at week 104 versus patients receiving golimumab from week 0; these patients also appeared to have more radiographic progression through week 104, This is consistent with observations through week 52,10 suggesting a delay in golimumab treatment may yield differences in disease activity persisting up to 2 years in favour of patients starting treatment earlier, although it is not known if those differences are clinically meaningful for treated patients.
The exact benefit of dose increase from golimumab 50 mg q4wks → 100 mg q4wks is difficult to discern given the lack of an adequate control arm. In general, there was no clear benefit of dose increase, with the exception of possible further improvement in the PASI score (median of 22% improvement in PASI score 12 weeks after dose escalation compared with before dose escalation).
Safety events observed through week 104 in these patients receiving golimumab after week 24 were consistent with safety reported through study weeks 24 and 52, with the exception of a case of histoplasmosis occurring after week 52 (golimumab 100 mg). Overall, 84% of golimumab-treated patients had ≥1 AE, with no clear difference between golimumab 50 and 100 mg in the frequency of AEs. Importantly, no patient developed active tuberculosis through 2 years, indicating that initiating treatment of latent tuberculosis prior to or concurrent with golimumab and close supervision of such patients during the treatment appear to be effective in decreasing the risk of developing active tuberculosis. Eight malignancies were observed in golimumab-treated patients, including four with BCC. The incidence of malignancies other than BCC was not different from the general US population per the SEER database.21 The observed incidence of NMSC through 2 years in golimumab-treated patients (four patients with BCC and baseline psoriasis) was within ranges observed in adalimumab psoriasis clinical trials.23
A challenge of conducting clinical trials in patients with chronic arthropathies (eg, PsA, RA) is the ethical concern of treating patients with placebo or suboptimal treatment for ≥3–6 months, yielding a paucity of long-term controlled data assessing risks/benefits of treatment versus control. Notwithstanding this deficiency, long-term uncontrolled data do provide additional insight. To mitigate the shortcomings of data collection in the GO-REVEAL long-term extension, efficacy data were analysed conservatively using an ITT approach and imputation for missing data and also considering patients who discontinued treatment due to lack of efficacy as ACR and DAS non-responders. Also, selected safety data such as malignancies were compared with a publically available database. These findings, although limited by the long-term extension design, support the long-term clinical and radiographic efficacy of golimumab treatment, as well as similar safety profiles between both golimumab doses and other anti-TNF PsA therapies. The data also suggest that both golimumab doses (50 and 100 mg), when administered q4wks, have similar efficacy and safety profiles and that increasing the dose from 50 to 100 mg q4wks adds limited benefit.
Acknowledgments
The authors thank Michelle Perate, MS, and Mary Whitman, PhD (Janssen Biotech, Inc), for writing and editorial support. A complete list of study investigators was previously reported.9
References
Footnotes
Handling editor Tore K Kvien
-
Contributors DDG participated in the trial design and conduct and manuscript preparation and has received grant support, consultation fees and/or honoraria from Abbott, Amgen, Bristol-Myers Squibb, Genentech, Janssen, Merck/Schering-Plough and Wyeth. AK participated in the trial design and conduct and manuscript preparation and has received funding for clinical research sponsored by Abbott, Amgen, Janssen and UCB. GGK participated in the trial design and conduct and manuscript preparation and has received fees as a consultant and/or advisory board member for Abbott, Almirall, Alza, Amgen, Anacor, Astellas, Barrier Therapeutics, Boehringer Ingleheim, Bristol-Myers Squibb, CombinatoRx, Exelixis, Genentech, Genzyme, Isis, Janssen, L'Oreal, Lupin Limited, Magen Biosciences, MedaCorp, Medicis, Merck/Schering-Plough, Novartis, Nova Nordisc, Somagenics, theDerm.org, Synvista, Warner Chilcot, UCB, USANA Health Sciences and ZARS. GGK has received lecture fees from Abbott, Amgen, Astellas, Boehringer Ingleheim, Connetics, Janssen, National Psoriasis Foundation, The Foundation for Better Health Care and Warner Chilcot. PJM participated in the trial design and conduct and manuscript preparation and has received research grant support, consultation fees and speaker honoraria from Abbott, Amgen, Biogen-IDEC, Bristol-Myers Squibb, Genentech, Janssen, Pfizer, and UCB, and grant support and consultation fees from Celgene and Novartis. IBM participated in the trial design and conduct and manuscript preparation and has received grant funding and honoraria from Abbott, Janssen, Roche, Merck/Schering-Plough and Wyeth. DvdH participated in the trial design, evaluation of radiographic assessments and manuscript preparation and has received consulting fees and/or research grants from Abbott, Amgen, AstraZeneca, BMS, Chugai, Eli-Lilly, GSK, Janssen, Novartis, Otsuka, Pfizer, Roche, Sanofi-Aventis, Merck/Schering-Plough, UCB and Wyeth. SM, WX and MM (data analysis, manuscript preparation) and AB (study design and conduct, manuscript preparation) are employees of Janssen Research & Development, LLC, a Johnson & Johnson (J&J) pharmaceutical company. ZX (data analysis, manuscript preparation) is an employee of J&J.
-
Funding The study was supported by Janssen Research & Development, Spring House, PA and Merck/Schering-Plough Corporation, Kenilworth, NJ.
-
Competing interests GGK participated in the trial design and conduct and manuscript preparation and has received fees as a consultant and/or advisory board member for Abbott, Amgen, Boehringer Ingleheim, Genentech, Janssen, L'Oreal, MedaCorp, Medicis, Merck/Schering-Plough and Novartis. GGK also has received lecture fees from Abbott, Amgen, Astellas, Boehringer Ingleheim, Janssen, National Psoriasis Foundation and The Foundation for Better Health Care.
-
Ethics approval Institutional review board or ethics committee approval and patient written informed consent were obtained prior to study participation.
-
Provenance and peer review Not commissioned; externally peer reviewed.