Article Text

Abstract
Objectives Osteoarthritis (OA) has a complex aetiology with a strong genetic component. Genome-wide association studies implicate several nuclear genes in the aetiology, but a major component of the heritability has yet to be defined at the molecular level. Initial studies implicate maternally inherited variants of mitochondrial DNA (mtDNA) in subgroups of patients with OA based on gender and specific joint involvement, but these findings have not been replicated.
Methods The authors studied 138 maternally inherited mtDNA variants genotyped in a two cohort genetic association study across a total of 7393 OA cases from the arcOGEN consortium and 5122 controls genotyped in the Wellcome Trust Case Control consortium 2 study.
Results Following data quality control we examined 48 mtDNA variants that were common in cohort 1 and cohort 2, and found no association with OA. None of the phenotypic subgroups previously associated with mtDNA haplogroups were associated in this study.
Conclusions We were not able to replicate previously published findings in the largest mtDNA association study to date. The evidence linking OA to mtDNA is not compelling at present.
- Gene Polymorphism
- Osteoarthritis
- Pharmacogenetics
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA) is the most common age-related joint disease. The pathogenesis of OA is complex, with several environmental and genetic factors implicated in the aetiology.1 Genome-wide association studies implicate several nuclear genes in the risk of developing OA, but a major component of the heritability remains unexplained.2
Mitochondria are intracellular organelles involved in the synthesis of ATP, the principal source of energy within cells. Several lines of evidence implicate mitochondrial dysfunction in the pathogenesis of OA, including the disruption of respiratory chain activity in chondrocytes,3 the increased production of reactive oxygen species which compromise chondrocyte function4 ,5 and a central role in one apoptotic pathway.6
Mitochondria contain multiple copies of the 16.5 kb mitochondrial genome (mitochondrial DNA (mtDNA)). MtDNA codes for 13 essential respiratory chain proteins and the RNAs required for intramitochondrial protein synthesis. Point mutations of mtDNA compromise oxidative phosphorylation and are a major cause of human disease.7 This raises the possibility that more subtle polymorphic variants contribute to genetic susceptibility of common complex traits, including OA. Being strictly maternally inherited, mtDNA undergoes negligible intermolecular recombination. Specific sequence variants that occurred 10–40 000 years ago define clades of the European mtDNA phylogeny called mtDNA haplogroups, each containing both distinct and shared genetic variants.8 Two studies have reported an association between different mtDNA haplogroups and OA, with one describing a reduced risk of knee OA with haplogroups J (defined by m.4216T>C and m.10398A>G) and JT (m.4216T>C), and another describing a reduced risk of hip OA with haplogroups J (defined by m.4216T>C and m.10398A>G) and J1c (m.14798T>C).9 ,10 However, each study lacked the power and resolution to reliably detect an association with all but the most common haplogroups. Moreover, despite being from the same geographic location, the studies reported different haplogroup associations with different joint involvement in OA for specific genders.9 ,10 While these data support the potential role of mtDNA in determining OA risk, the absence of direct replication means that the role of mtDNA in OA is thus uncertain, and the joint-specific risk is difficult to explain based on the proposed mechanism. To address this issue, we carried out a two phase study of 138 mtDNA variants in 7393 OA cases and 5122 controls as part of the arcOGEN consortium.
Methods
We performed a two stage genetic association study. The cases were part of the arcOGEN study, and the controls were part of the Wellcome Trust Case Control Consortium 2 (WTCCC2) study, both ascertained as described previously.2 ,11 In cohort 1, 3093 cases (ARC1) were compared with 2640 controls from the 1958 Birth Cohort (WTCCC-58C). In cohort 2, 4300 cases (ARC2) were compared with 2482 UK Blood Service controls (WTCCC-NBS). All cases were genotyped using the Illumina Human610 platform (Illumina, San Diego, California, USA). All controls were genotyped on the Illumina 1.2M Duo platform (Illumina). Individuals were excluded from subsequent analysis if data were absent in >10% of single nucleotide polymorphism (SNP) (cohort 1 cases=0 and WTCCC-58C=44 and cohort 2 cases=0 and WTCCC-NBS=11) and SNPs were excluded if >10% of genotypes were absent (cohort 1=5 and cohort 2=4).11 We excluded 30 SNPs (cohort 1=15 and cohort 2=21) with a study-wide missing data rate >5% or >1% for SNPs with a study-wide minor allele frequency (MAF) <5%.11 Finally, 64 SNPs with MAF<1% were removed (cohort 1=53 and cohort 2=64). Subsequent analysis was restricted to a concordant dataset of 62 mtDNA variants passing quality control (QC) in both phases. Differential missingness tests between cases and controls revealed significant differences in 14 SNPs (p=<10−4) reducing the final number of experimental SNPs to 48. Statistical significance was defined as p<0.05 in both phases. χ² And missingness were computed using PLINK v 2.050 (http://pngu.mgh.harvard.edu/purcell/plink/).12
Array genotypes were used to identify mtDNA haplogroup-specific sequence motifs, when compared with the mtDNA reference sequence (see online supplementary materials and table S1), allowing 98.48% of subjects to be successfully assigned to a European haplogroup.
A principal components analysis (PCA )13 was performed on the X : Y ratios of raw intensities for all SNPs for the combined dataset of cases (n=7393) and controls (n=5122) using R. The k-means function of the R cluster package was used to cluster individuals into 5, 10, 15 and 20 groups using these PCA scores.
Results
Using established QC criteria14 we rejected 90 SNPs, leaving 48 in both cohorts. There was no association between any mtDNA variant passing QC and OA. Stratifying by gender, joint involvement (knee or hip) and method of case ascertainment (radiograph or arthroplasty) failed to reveal any significant associations (see online supplementary table S2).
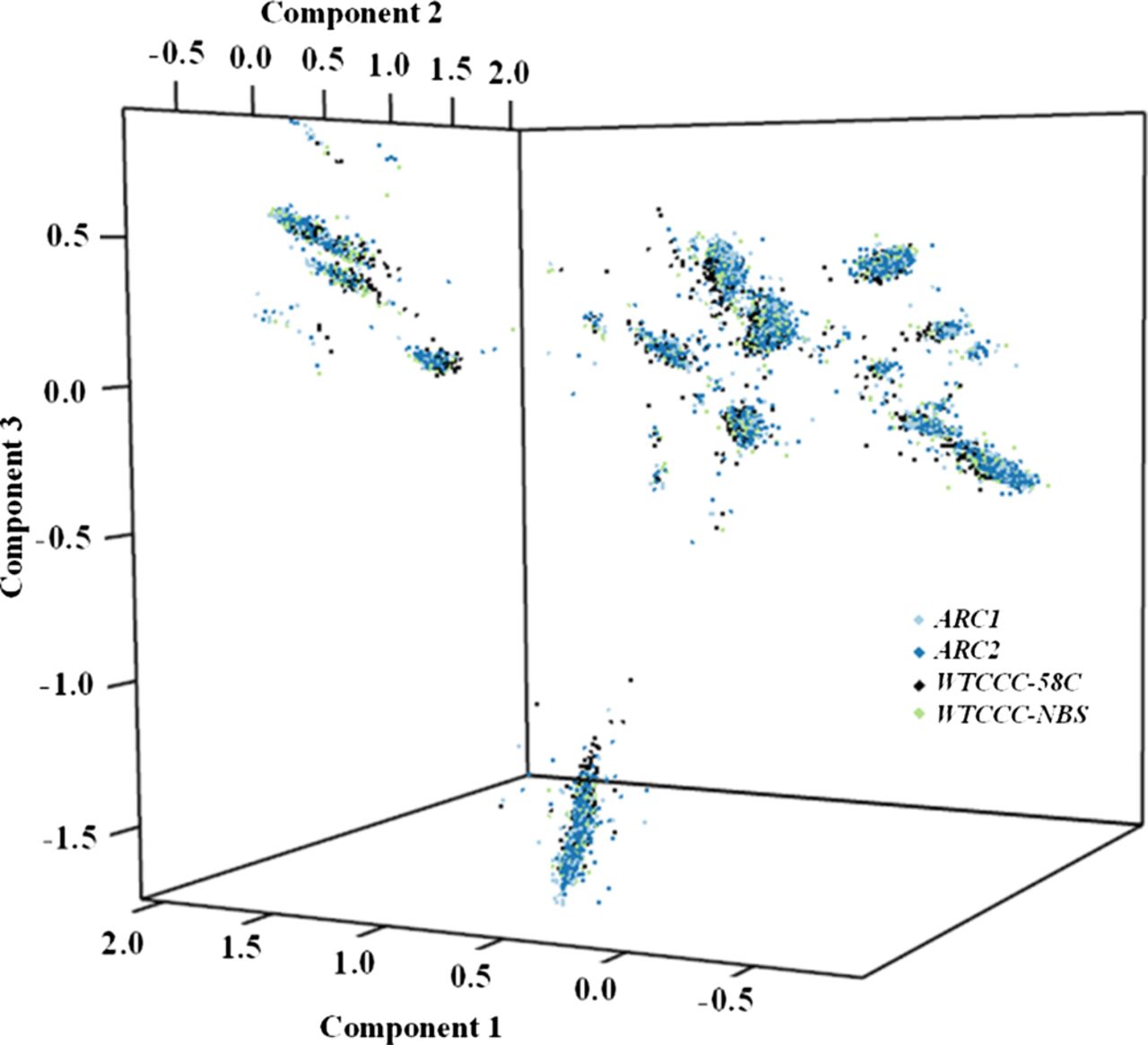
We then studied mtDNA haplogroups using two independent approaches: (1) manual haplogroup calling based on phylogenetics (for methods see online supplementary table 1) and (2) hypothesis-free approach based on raw fluorescence intensities in a principal components analysis. Analysis of mtDNA haplogroups between cases and controls failed to replicate previous studies (table 1). Stratifying by gender, joint involvement (knee or hip) and method of case ascertainment (radiograph or arthroplasty) failed to reveal any significant associations with mtDNA haplogroups (see online supplementary table S3). PCA analysis identified the main mtDNA haplogroups previously identified through phylogenetic analysis. These appear as clusters in figure 1, where each cluster represents a major European mtDNA haplogroup. However, there was no significant difference in cluster membership for any of the disease or control cohorts, or when the cases and controls were pooled (figure 1, online supplementary table S4). The data were only pooled after confirming that the allele frequencies were not significantly different between the two case groups or between the two control groups.
Frequency of mitochondrial DNA (mtDNA) haplogroups in osteoarthritis and control subjects
{kind=link}
Principal components analysis (PCA) performed on raw fluorescent intensities in the pooled data set of 7393 osteoarthritis (OA) cases and 5122 controls. The PCA provided independent confirmation of the European haplogroup structure. There was no significant difference in the cluster distribution between OA cases and controls (online supplementary table S4). 58C, WTCCC MRC 1958 birth cohort control genotypes; ARC1, arcOGEN cohort 1; ARC2, arcOGEN cohort 2; NBS, WTCCC national blood transfusion service control genotypes; WTCCC, Wellcome Trust Case Control Consortium. This figure is only reproduced in colour in the online version.
Discussion
The previously reported haplogroup associations were with specific subgroups of OA patients, subcategorised based on gender and/or specific joint involvement.9 ,10 Our study had >99% power to detect these associations, and there was no significant difference in the frequency of the previously associated SNPs between controls in the published studies9 ,10 and the controls used in our study. The previously published results were based on substantially smaller study groups than the ones we describe here, raising the possibility that the published findings are a false positive finding. On the other hand, the relative contribution of specific mtDNA variants could vary in different ethnic groups, possibly through an interaction with environmental factors and different nuclear genes.15 In practice, this means that the specific mtDNA variants which fail to show an association with disease in this study could be associated with disease in a different ethnic population. Geographic variation in allelic association could also arise through homoplasy. Homoplasy is the recurrence of mutations on different branches of the mtDNA phylogeny in different parts of the world. Homoplasy accounts for up to 20% of mtDNA variation, and often involves non-synonymous substitutions.16 This raises the possibility that haplogroup markers tag different homoplastic functional variants in different populations. If the homoplasies are having a functional effect, then this would lead to different haplogroup associations in different studies across the globe. Finally, it is possible that geographic differences in the fine detail of the sub-haplogroup structure of mtDNA could account for inconsistencies between studies, as described for Leber's hereditary optic neuropathy, where sub-branches of haplogroup J are associated with either an increased or decreased risk of visual failure in different populations, largely due to specific differences in the cytochrome B protein sequence.17 However, in the largest mtDNA association study to date, we found no association between OA and the major European mtDNA haplogroups in either cohort. If an association does exist between OA and mtDNA, it is likely that this will only be resolved through extensive genotyping, ideally at the whole mtDNA level, in a much larger cohort of cases and controls and should be consistent in more than one geographic region.
Acknowledgments
The UK samples used in arcOGEN derive from eight centres: Nottingham, London, Oxford, Sheffield, Southampton, Newcastle, Wansbeck and Edinburgh. For Nottingham we acknowledge Arthritis Research UK for funding the collection of the majority of extant cases (cohort 1) and we thank Sally Doherty and Maggie Wheeler for organising cohort 2 patient ascertainment. For London we thank the staff from the TwinsUK unit and the Chingford Study for cohort 1 patient ascertainment, we thank Fiona O'Neill for organising cohort 2 patient ascertainment, and we acknowledge financial support from Arthritis Research UK, from the Wellcome Trust, from the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre (BRC) award to Guy's & St Thomas' NHS Foundation Trust in partnership with King's College London, and from the European Commission framework 7 programme TREAT-OA (grant 200800). For Oxford we acknowledge funding support from the Collisson Foundation, the Botnar Foundation and the Jean Shanks Foundation for cohort 1 patient ascertainment, we acknowledge the NIHR for supporting the Biomedical Research Unit (BRU) at the University of Oxford, and we thank Bridget Watkins, Anne Hanley and Kim Clipsham for cohorts 1 and 2 patient ascertainment. For Sheffield we acknowledge the NIHR for supporting the Sheffield Musculoskeletal BRU collection site, the South Yorkshire Clinical Research Network for part-funding the Sheffield research nurses and for clerical support, the Royal College of Surgeons of England and the Cavendish Hip Foundation, Andrew Gordon for cohort 1 patient ascertainment, and Diane Swift, Ellen Paling and Adam Taylor for cohort 2 patient ascertainment. For Southampton we acknowledge the Wellcome Trust Clinical Research Facility at Southampton General Hospital, we thank Phillippa-Kate Battley and Elizabeth Arden for organising cohort 2 patient ascertainment, and we thank Richard Keen and Anna Bara for facilitating access to the Arthritis Research UK-funded VIDEO study. For Newcastle we acknowledge Sheryl Mitchell and the Freeman Hospital arthroplasty surgeons Nigel Brewster, David Deehan, Craig Gerrand, Andy Gray, Munawar Hashmi, Jim Holland and David Weir for assistance in cohort 2 patient ascertainment, we thank Karen Bettinson for her assistance, we acknowledge the support of the UK NIHR BRC for Ageing and Age-related disease award to the Newcastle upon Tyne Hospitals NHS Foundation Trust and the support of the Northumberland, Tyne and Wear CLRN. For Wansbeck we thank Kirsten Walker and Donna Gray for organising cohort 2 patient ascertainment, and we acknowledge the Northumbria NHS Foundation Trust arthroplasty consultants Mr Partington, Mr Muller, Mr Emmerson, Mr Prasad, Mr Kramer, Mr Jones, Mr Murty, Mr Asaad, Mr Leitch, Mr Pratt, Mr Townshend and Miss Van Kampen for accrual. For Edinburgh we acknowledge the contribution of Nichola Koller, Gayle Scott and Helen Watters for recruitment and ascertainment of cohort 2 cases. We acknowledge cohorts 1 and 2 sample management undertaken by the UK DNA Banking Network funded by the Medical Research Council at CIGMR—the Centre for Integrated Genomic Medical Research, University of Manchester, with special thanks to Kate Sherburn and Kate Dixon. Genotyping was performed at the Wellcome Trust Sanger Institute and we thank Emma Gray, Sarah Edkins and Cordelia Langford for their assistance. We thank Vanessa Cox of the MRC Epidemiology Resource Centre, Southampton, for database and computing services for the arcOGEN cohort 2 patient questionnaires.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributions GH and IW carried out the primary analysis, supervised by PFC, DCS and JL. KP, LS, NWR, NA, FB, IC, AC, KC, PD, MD, AMcC, WERO, SHR, MRR, TDS, AMV, GAW, JMW, EZ and JL generated the primary data. PFC, JL and GH wrote the manuscript, which was modified following comments from the other authors . The authors declare no conflict of interest.
-
Funding arcOGEN (http://www.arcogen.org.uk/) was funded by a special purpose grant from Arthritis Research UK (grant 18030). This study used genotype data from population controls that were generated by the Wellcome Trust Case Control Consortium 2 (a full list of the investigators who contributed to the generation of the data is available from http://www.wtccc.org.uk, funded by The Wellcome Trust (grant 083948). The population controls were from the 1958 British Birth Cohort collection funded by the Medical Research Council (grant G0000934) and The Wellcome Trust (grant 068545) and from the UK Blood Services Collection of Common Controls funded by The Wellcome Trust. KP, LS and EZ are supported by the Wellcome Trust (098051), KC is supported by a Botnar Fellowship, NWR is supported by the Wellcome Trust (WT079557MA) and JMW is supported by the Higher Education Funding Council for England. The funding sources were not involved in the study design; in the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the paper for publication. Funding for the project was provided by the Wellcome Trust under award 076113 and 085475. PFC is a Wellcome Trust Senior Fellow in Clinical Science (WT084980/Z/08/Z) and an NIHR Senior Investigator, who is also supported through the Wellcome Trust centre for Mitochondrial Research (WT096919Z/11/Z), the Medical Research Council (UK), the UK Parkinson’s Disease Society, and the UK NIHR Biomedical Research Centre for Ageing and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust.
-
Competing interest None.
-
Ethics approval Ethics approval was provided by Newcastle REC.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/