Article Text

Abstract
Objective To assess the efficacy and safety of golimumab + methotrexate (MTX) in Japanese patients with active rheumatoid arthritis (RA).
Methods 269 Japanese patients with active RA despite treatment with MTX were randomised (1:1:1) to placebo + MTX (Group 1), golimumab 50 mg + MTX (Group 2) or golimumab 100 mg + MTX (Group 3). Subcutaneous golimumab/placebo was injected every 4 weeks; stable doses of oral MTX (6–8 mg/week) were continued. Patients were allowed to enter early escape (Group 1 added golimumab 50 mg, Group 2 increased golimumab to 100 mg, Group 3 continued golimumab 100 mg) based on swollen/tender joint counts at week 14. The primary study endpoint was achievement of at least 20% improvement in the American College of Rheumatology (ACR20) response criteria at week 14. To control for multiplicity of testing, treatment group comparisons were first made between combined Groups 2 and 3 versus Group 1, followed by comparisons of Group 2 and Group 3 versus Group 1.
Results The proportion of patients with an ACR20 response at week 14 was significantly higher in combined Groups 2 and 3 (73.4%, 127/173) and in each of Group 2 (72.1%, 62/86) and Group 3 (74.7%, 65/87) compared with Group 1 (27.3%, 24/88; p<0.0001 for all comparisons). Golimumab + MTX also elicited a significantly better response than placebo + MTX in other efficacy parameters, including disease activity score (DAS28) response/remission and radiographic assessments. During the 16-week fixed treatment regimen study period, 72.7%, 75.6% and 78.2% of patients had adverse events and 1.1%, 1.2% and 2.3% had serious adverse events in Groups 1, 2 and 3, respectively.
Conclusion In Japanese patients with active RA despite MTX therapy, golimumab + MTX was significantly more effective than MTX monotherapy in reducing RA signs/symptoms and limiting radiographic progression with no unexpected safety concerns.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory disease mediated by overproduction of cytokines such as tumour necrosis factor α (TNF).1 2 Golimumab, a newer human anti-TNF monoclonal antibody that binds with high affinity and specificity to soluble and transmembrane TNF,3 antagonises the effects of TNF.1 Golimumab + methotrexate (MTX) has demonstrated statistically significant efficacy versus MTX monotherapy in MTX-naïve patients with RA4 and in patients with active RA despite prior MTX therapy.5 6
In a phase 1 study of healthy age- and dose-matched Japanese men (n=24) and Caucasian subjects (n=27), the pharmacokinetics of golimumab were comparable between ethnic groups.7 A phase 2/3 study was conducted to examine the efficacy and safety of golimumab in Japanese patients with active RA despite MTX therapy.
Methods
Patients
Eligible patients were adults (age 20–75 years) with RA diagnosed according to the American College of Rheumatology (ACR) 1987 revised criteria,8 with disease duration of ≥3 months who had received ≥6 mg/week oral MTX for RA for ≥3 months before study agent initiation. Stable MTX doses (6–8 mg/week) were required for ≥4 weeks before the start of the study. Patients had to have active RA (≥4/66 swollen joints and ≥4/68 tender joints at screening/baseline) and had to meet at least two of the following criteria at screening/baseline: (1) C-reactive protein (CRP) >1.5 mg/dl or erythrocyte sedimentation rate (ESR) by the Westergren method of >28 mm/h, (2) morning stiffness lasting ≥30 min, (3) radiographic evidence of bone erosion, or (4) anti-cyclic citrullinated peptide antibody-positive or rheumatoid factor-positive. Eligible patients also met prespecified concomitant medication and tuberculosis screening criteria (see online supplement).
Study design
This multicentre phase 2/3 study (ClinicalTrials.gov NCT00727987) had a 24-week, randomised, double-blind, placebo-controlled phase followed by an open-label extension continuing through 3 years. This report presents clinical data through week 24. The study was conducted according to Declaration of Helsinki and Good Clinical Practice guidelines. The protocol was reviewed and approved by all institutional review boards. All patients provided written informed consent prior to study participation.
Eligible patients were randomly (1:1:1) assigned to receive placebo injection + oral MTX (Group 1), golimumab 50 mg injection + oral MTX (Group 2) or golimumab 100 mg injection + oral MTX (Group 3). Golimumab and placebo were supplied as sterile liquid (Janssen Biotech Inc, Horsham, Pennsylvania, USA) for subcutaneous injection at week 0 and every 4 weeks to week 24. MTX doses were not adjusted unless dose reduction was required because of MTX toxicity.
At week 16, patients with <20% improvement from baseline in tender and swollen joint counts at week 14 could enter double-blind early escape (EE). Group 1 added golimumab 50 mg, Group 2 increased the golimumab dose to 100 mg and Group 3 continued golimumab 100 mg.
Study endpoints
The primary study endpoint was response according to achievement of at least 20% improvement in the ACR response criteria9 at week 14, prior to any change in treatment at week 16. Additional efficacy assessments included ACR50 and ACR70 responses, ACR-N Index of Improvement10 and Disease Activity Score using 28 joints and ESR (DAS28(ESR)). DAS28(ESR) response (moderate and good ratings) and remission (DAS28(ESR) score <2.6) were also determined.11 12 Physical function was assessed using the disability index of the Health Assessment Questionnaire (HAQ-DI).13 All efficacy assessments were conducted at baseline (week 0) and at weeks 4, 8, 12, 14, 16, 20 and 24.
Hand and feet x-rays were obtained before administration of study agent at weeks 0 and 24 or upon premature discontinuation. They were scored by the BioClinica Corporation (Newtown, Pennsylvania, USA) using the Sharp score as modified by van der Heijde and colleagues (vdH-S).14 Two primary readers who were blinded to patient identity, treatment group assignment and x-ray time point read the x-rays. If the readers' scores differed by ≥10 points or data were unavailable for one reader, a third reader evaluated the x-rays. In the former case, the reader score that differed the least from the adjudicator's score was used.
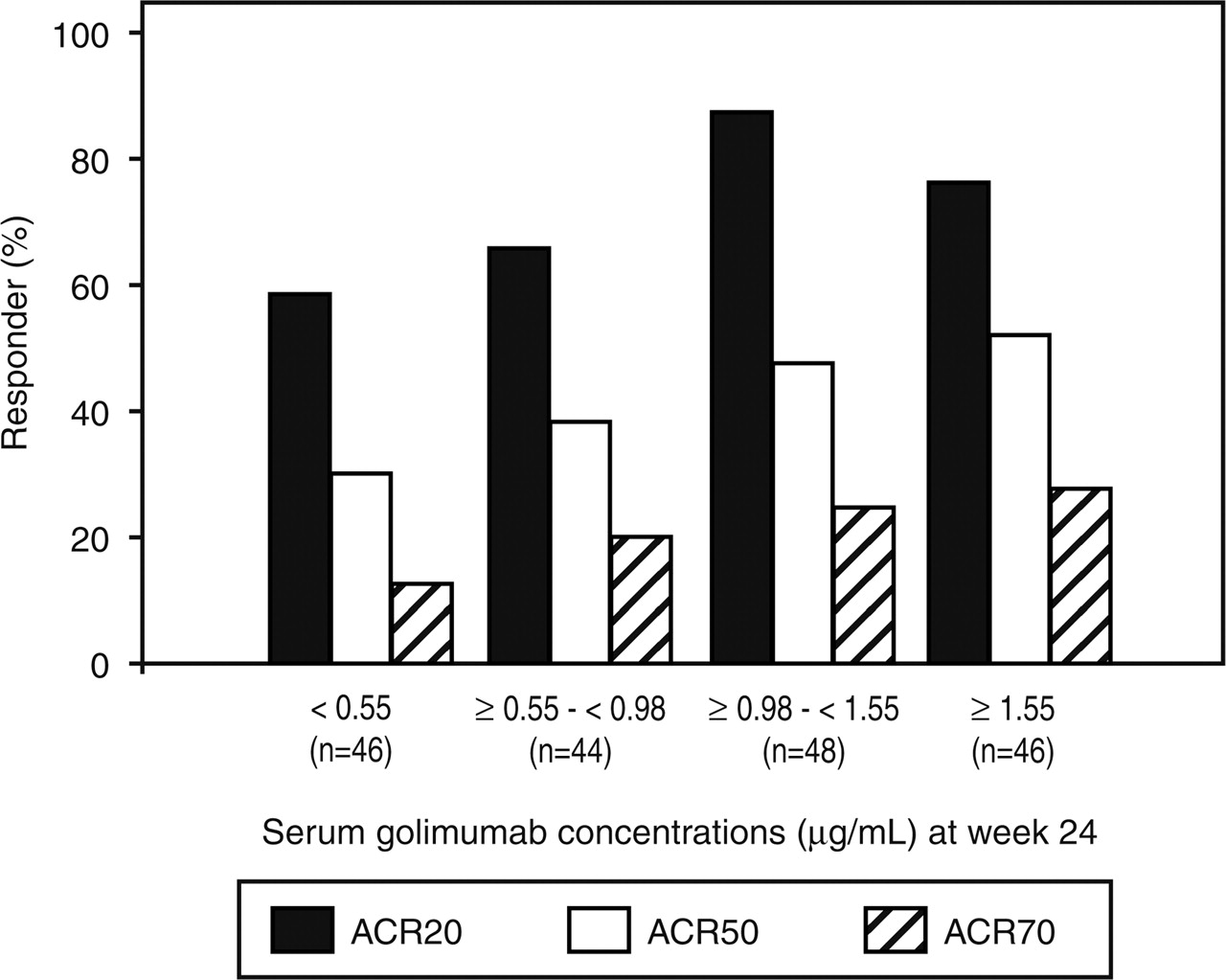
In a post hoc analysis, the relationship between efficacy and serum study agent concentrations was examined, whereby ACR response rates were categorised by serum golimumab concentration quartiles: <0.55 µg/ml (n=46), ≥0.55–<0.98 µg/ml (n=44), ≥0.98–<1.55 µg/ml (n=48) and ≥1.55 µg/ml (n=46).
Safety assessments included adverse events (AEs) and routine laboratory analyses. Serum golimumab concentrations and antibodies to golimumab were determined.15
Statistical analyses
Efficacy and pharmacology parameters were primarily assessed according to a modified intent-to-treat approach in which patients who did not meet the study eligibility criteria, did not receive study treatment and/or had no efficacy- or pharmacology-related data following randomisation were excluded from the full analysis patient population. Safety analyses included all randomised treated patients. Further details of prespecified data handling rules and sample size calculations are provided in the online supplement.
Treatment group differences in dichotomous variables were assessed with a χ2 test. Type I error at the 0.05 level of significance was preserved with a hierarchical approach to control for multiplicity when testing, wherein the comparison between combined Groups 2 and 3 versus Group 1 was made first. If this difference was significant, pairwise comparisons between Group 2 versus Group 1 and Group 3 versus Group 1 were performed. In data summaries that did not present patients who entered EE separately, such patients were grouped by randomised group and had week 24 data replaced with week 16 data. For continuous variables, treatment group differences were assessed using analysis of covariance (ANCOVA) with treatment as a factor and baseline value as a covariate or analysis of variance (ANOVA) with treatment as a factor. For comparisons of changes in vdH-S score, ANCOVA based on least squares mean and accompanying two-sided 95% confidence intervals was detailed a priori, and ANOVA based on van der Waerden normal scores was conducted post hoc for ease of comparison with the radiographic results of the GO-FORWARD study.16 ANCOVA results are presented herein. A cumulative probability plot depicting changes in the vdH-S score (shown in ascending order of magnitude with smaller changes indicating greater inhibition of disease progression) was also constructed. The proportions of patients with no change in the vdH-S score and with changes in excess of the smallest detectable change (SDC=3.23) were also determined and compared among treatment groups with a χ2 test. Agreement between the two primary readers for vdH-S scores was assessed by determination of intraclass correlation coefficients (ICCs).
Results
Patient disposition and baseline characteristics
Data for this report were collected beginning in May 2008 and the week 24 database was locked in September 2009. Two hundred and sixty-nine patients were enrolled at 89 investigational sites in Japan and randomised to Group 1 (n=90), Group 2 (n=89) or Group 3 (n=90); 261 patients received at least one study treatment (n=88, 86 and 87 in Groups 1, 2 and 3, respectively). Eight patients discontinued the study before receiving study treatment. Similar proportions of treated patients completed subcutaneous administration of the study agent through the week 24 visit in Group 1 (95.5%), Group 2 (94.2%) and Group 3 (92.0%) (figure 1).
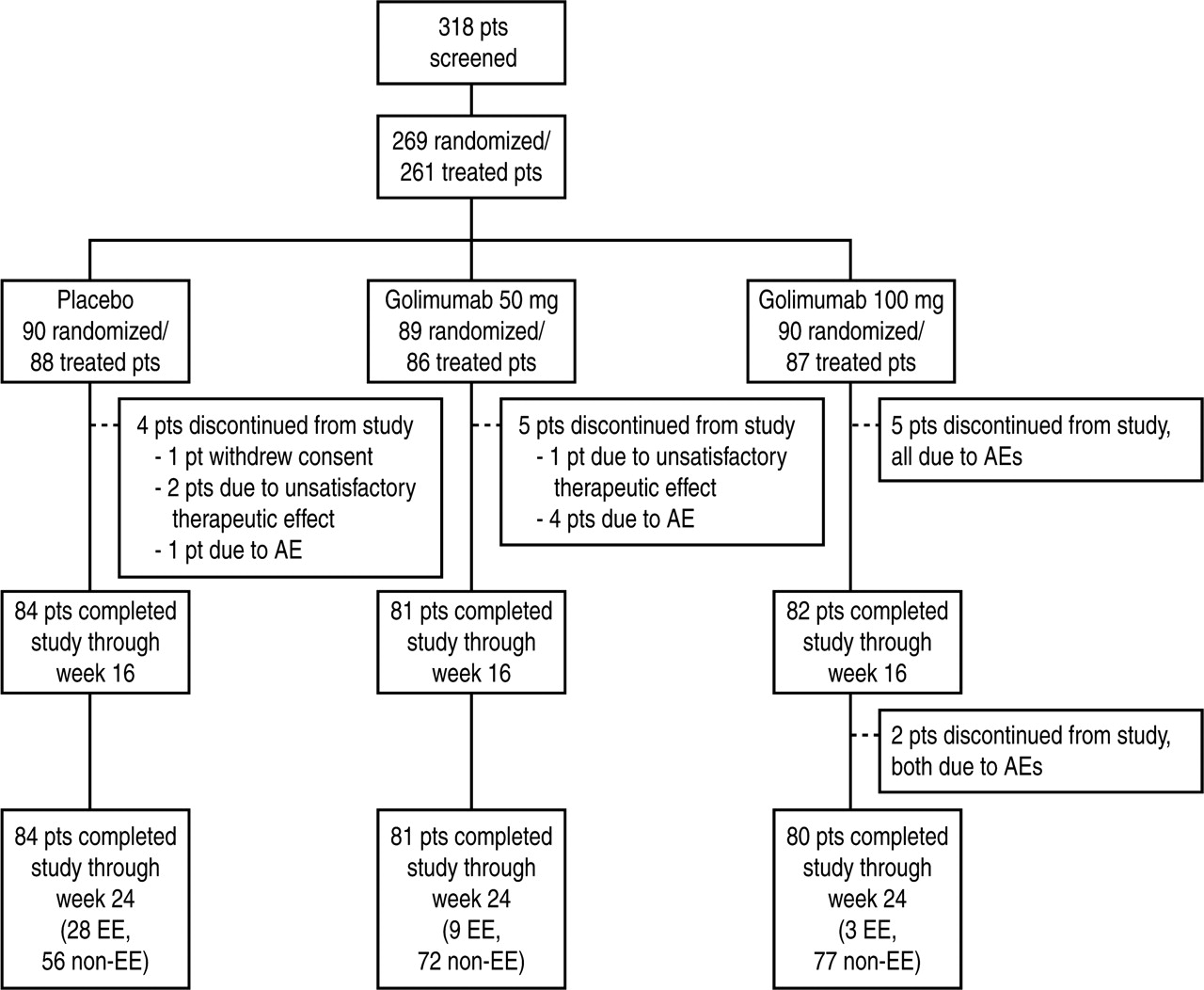
Patient disposition through week 24; randomised patients. Note that ‘worsening of rheumatoid arthritis’ is included in ‘unsatisfactory therapeutic response’ and not as an AE. AE, adverse event; EE, early escape; pts, patients.
The overall mean (SD) baseline vdH-S score was 55.1 (58.1) and duration of RA was 8.5 (7.9) years. Baseline demographic and disease characteristics were generally consistent across the three treatment groups, with the exception of shorter mean disease duration (8.1 years) and lower mean baseline CRP level (1.5 mg/dl) in Group 3 compared with Group 1 (8.7 years and 2.2 mg/dl, respectively) and Group 2 (8.8 years and 1.9 mg/dl, respectively) (table 1).
Baseline patient and disease characteristics: full analysis patient population*
Efficacy results
ACR response
Analysis of the primary endpoint (ie, ACR20 response at week 14) demonstrated a significant difference between combined Groups 2 and 3 (73.4%, 127/173) and Group 1 (27.3%, 24/88) (p<0.0001; table 2). Significantly higher ACR20 response rates were also observed in Group 2 (72.1%, 62/86; p<0.0001) and Group 3 (74.7%, 65/87; p<0.0001) versus Group 1. Consistent findings were observed for ACR50 and ACR70 responses (table 2).
Summary of clinical and radiographic efficacy at weeks 14 and 24: full analysis patient population*
Differences in ACR response between golimumab + MTX and placebo + MTX were evident as early as week 4 and maintained through week 24 (figure 2). Patients in Group 1 who crossed over to golimumab 50 mg + MTX and patients in Group 2 who increased the golimumab dose from 50 mg to 100 mg + MTX appeared to demonstrate clinical benefit following the change in study treatment (figure 2).

(A) American College of Rheumatology 20% (ACR20), (B) 50% (ACR50) and (C) 70% (ACR70) improvement from baseline through week 24. Note that patients who met the early escape criteria at week 16 and crossed over to golimumab 50 mg or dose escalated from golimumab 50 mg to 100 mg are shown with an open triangle and closed circle, respectively. For the 28 patients in the placebo + MTX group and the nine patients in the golimumab 50 mg + MTX group who met the early escape criteria, week 20 and 24 data were imputed using last observation carried forward methodology, as were other missing data. As such, 88 patients in the placebo + MTX group and 86 patients in the golimumab 50 mg + MTX group were included in these data displays. MTX, methotrexate.
Other clinical measures of RA and physical function
Statistical comparisons of combined Groups 2 and 3 versus Group 1, as well as for Group 2 versus Group 1 and Group 3 versus Group 1, were significant for supportive clinical efficacy parameters including ACR-N Index of Improvement, DAS28(ESR) response and DAS28(ESR) remission (table 2). At week 14, a significantly greater median improvement in the HAQ-DI score was observed in patients who received golimumab + MTX (median of 0.25 for combined Groups 2 and 3, Group 2 and Group 3) versus placebo + MTX (median 0.13; p<0.0001 for all comparisons). Improvements in the HAQ-DI score at week 24, as well as the proportions of patients achieving a HAQ score <0.5, were also significantly greater among patients who received golimumab + MTX versus placebo + MTX (table 2).
Radiographic progression
The primary readers exhibited good agreement with regard to vdH-S scores, with ICCs of 0.98 for baseline scores, 0.98 for week 24 scores and 0.80 for the change from baseline to week 24 in vdH-S scores.
Significantly less radiographic progression from baseline to week 24 was observed in patients who received golimumab + MTX (median changes in total vdH-S score of 0.00 (p=0.0009) for combined Groups 2 and 3, 0.00 (p=0.0203) for Group 2 and 0.00 (p=0.0006) for Group 3) versus placebo + MTX (median change 0.25). Treatment group differences in the total vdH-S score were largely attributable to significantly less change in the erosion score with golimumab + MTX therapy. As shown in the cumulative probability plot shown in figure 1 in the online supplement, changes in vdH-S scores were smaller and thus inhibition of radiographic progression was greater in patients treated with golimumab + MTX (Group 2 and Group 3) than in those given placebo + MTX (Group 1).
Significantly greater proportions of patients in combined Groups 2 and 3 (64.7%, p=0.0217) and Group 3 (70.1%, p=0.0066) did not have an increase in the total vdH-S score (ie, change from baseline to week 24 <0) compared with Group 1. The proportions of patients with a change in the total vdH-S score from baseline to week 24 greater than the SDC (3.23) were also significantly lower in combined Groups 2 and 3 (11.0%, p=0.0216) and Group 3 (5.7%, p=0.0023) compared with Group 1 (table 2).
Golimumab pharmacokinetics and antibodies to golimumab
Median serum golimumab concentrations were approximately dose proportional and appeared to have reached steady state by week 14. Median serum golimumab concentrations at weeks 12 and 16 were 0.72 and 0.73 µg/ml, respectively, for Group 2 and 1.28 and 1.16 µg/ml, respectively, for Group 3. These steady state concentrations were maintained at week 24. In Group 2, serum golimumab concentrations in patients who met the EE criteria were approximately 45–82% of those in Group 2 patients who did not meet the EE criteria (data not shown).
In an analysis of week 24 ACR response by week 24 golimumab concentration quartiles, the lowest response rates occurred in patients with serum golimumab concentrations <0.55 µg/ml, followed by concentrations ≥0.55–<0.98 µg/ml (figure 3). No patient developed antibodies to golimumab.

{kind=link}
{kind=link}
{kind=link}
Proportions of patients achieving at least 20%, 50% and 70% improvement in the American College of Rheumatology (ACR20, ACR50, ACR70) response criteria by serum golimumab concentration quartiles (µg/ml) at week 24. The results are from a post hoc analysis of ACR responders in the combined Group 2 (golimumab 50 mg + MTX) and Group 3 (golimumab 100 mg + MTX). MTX, methotrexate.
Adverse events
AEs reported at week 16 (fixed treatment regimen study period) and week 24 are summarised in table 3. By week 16, 72.7% (64/88), 75.6% (65/86) and 78.2% (68/87) of patients in Groups 1, 2 and 3, respectively, had AEs. Infections were the most common AEs in Group 1 (35/88, 39.8%), Group 2 (33/86, 38.4%) and Group 3 (29/87, 33.3%) through week 16 and were also the most common AEs at week 24 (table 3).
Summary of safety through weeks 16 and 24 in all randomised patients who received at least one injection of study agent
Serious AEs were relatively uncommon through week 16, occurring in one patient (1.1%) in Group 1 (intervertebral disc protrusion), one patient (1.2%) in Group 2 (ileus) and two patients (2.3%) in Group 3 (herpes zoster/tendon rupture and aortic dissection). Two additional patients had serious AEs between weeks 16–24, including bone neoplasm (thoracic vertebra tumour (haemangoendothelioma) with ‘borderline’ or low malignancy potential) in Group 2 and humeral fracture/cruciate ligament injury in Group 3, yielding a total of five (2.5%) patients treated with golimumab + MTX with serious AEs through week 24. No deaths or malignancies were reported.
In addition, by week 16, one (1.1%), three (3.5%) and six (6.9%) patients in Groups 1, 2 and 3, respectively, discontinued the study agent because of an AE. By week 24, 11 (5.5%) of the 201 patients treated with golimumab + MTX had discontinued golimumab due to AEs; these included infection (n=2), skin disorders (n=2), liver function abnormality (n=2), injury (n=2), bone neoplasm (n=1), aortic dissection (n=1), gastrointestinal disorder (n=1) and elevated blood pressure (n=1 in combination with skin disorder).
As noted, infection was the most common system organ class of AEs, occurring in 35 (39.8%), 33 (38.4%) and 29 (33.3%) patients in Groups 1, 2 and 3, respectively, up to week 16. By week 24, 74 (36.8%) patients treated with golimumab + MTX had an infection, most commonly rhinopharyngitis (19.4%, 39/201), gastroenteritis (3.5%, 7/201) and pharyngitis (3.0%, 6/201). No patient developed tuberculosis.
Injection site reactions were reported in six (6.8%), seven (8.1%) and nine (10.3%) patients in Groups 1, 2 and 3, respectively, up to week 16. By week 24, 10.4% (21/201) of all patients treated with golimumab + MTX had an injection site reaction. Erythema at the injection site was the most common of these AEs. All injection site reactions were considered mild and none required cessation of the study agent. No cases of anaphylactic reaction or serum sickness-like reactions were observed.
Discussion
This study evaluated the efficacy of golimumab 50 mg and 100 mg administered subcutaneously every 4 weeks in combination with MTX (6–8 mg/week) versus MTX (6–8 mg/week) monotherapy in Japanese patients with active RA despite MTX therapy. A significantly higher proportion of patients randomised to golimumab 50 mg or 100 mg + MTX (combined Groups 2 and 3) achieved an ACR20 response at week 14 than those receiving MTX monotherapy (73.4% versus 27.3%; p<0.0001). Significantly higher ACR20 response rates were also observed for the individual golimumab dose groups. While the primary endpoint at week 14 did not coincide with trough golimumab concentrations, ACR20 response rates at the time of trough concentrations (week 16) were comparable to those observed at week 14 (ie, 71.7% and 29.5%, respectively, in combined Groups 2 and 3 and Group 1, respectively; data not shown).
These primary endpoint results were consistent with the results of the GO-FORWARD study, a large phase 3 multicentre trial of golimumab encompassing a similar design (primary endpoint at week 14 and treatment change due to EE from week 16 onwards) and a comparable population of patients with RA (approximately 15% of whom were Asian; data on file, Centocor Research & Development) with an inadequate response to MTX.5 Consistency between our findings and those of the GO-FORWARD study was also observed for improvements in HAQ-DI at week 24.5
Significantly less radiographic progression was observed at week 24 with golimumab + MTX than with placebo + MTX, and findings of a post hoc ANOVA analysis of vdH-S scores based on the van der Waerden normal scores were consistent (data not shown). In the GO-FORWARD study, however, minimal radiographic progression was observed in all treatment groups during the same time period, yielding no significant differences between golimumab + MTX and placebo + MTX.5 16 Minimal radiographic progression was probably related to minimal baseline active inflammation (median CRP 0.8–1.0 mg/dl).5 16 In a separate study of golimumab, MTX-naïve patients with RA had higher baseline CRP levels (median 1.3–1.4 mg/dl), greater radiographic progression than in the GO-FORWARD study despite less baseline radiographic damage and significantly less radiographic progression at week 28 with golimumab + MTX versus placebo + MTX.5 16 Thus, CRP is likely to be a more important predictor of radiographic progression than the baseline radiographic score since radiographic progression is less likely if there is no active inflammation, regardless of the amount of baseline radiographic damage.16 The CRP concentration has also been shown to predict ACR20 response.17 In this context, the participants in the current study had an intermediate amount of active inflammation at baseline (median CRP 0.9–1.3 mg/dl) and also demonstrated significantly less radiographic progression at week 24 with golimumab + MTX compared with placebo + MTX. In evaluating the radiographic data, it is important to note that the statistically significant differences between the groups are driven by a subset of patients who progress more rapidly than the overall population, and it is in those patients that the treatment effect becomes clinically relevant.
Of note, the MTX dose used in this trial, while consistent with that approved in Japan at the time the trial was planned, was suboptimal (6–8 mg/week) in the context of customary doses elsewhere18 and as used in the GO-FORWARD study (15–25 mg/week).16 Evaluation of the efficacy and safety of MTX doses >8 mg/week in Japanese patients with RA has yielded a favourable benefit/risk profile19 and approved dosing is now extended to up to 16 mg/week. It would therefore be prudent to reassess the responses to golimumab as approved MTX doses in Japan are harmonised with those approved in North America and Europe for RA. These suboptimal MTX doses may explain the higher ACR20 response rates observed in the current golimumab trial (∼70%) compared with previously conducted trials of golimumab in RA (~60%) in which more robust ongoing MTX treatment regimens (10–15 mg/week) could have resulted in less room for improvement from baseline.4 5 It is noteworthy that, when assessing response according to the more stringent ACR50 and ACR70 response criteria, the background MTX dose does not appear to affect the clinical response.4 5 Similar reasoning may be applied to explain the highly significant difference in radiographic progression observed between placebo + MTX and golimumab + MTX despite only an intermediary level of baseline inflammation compared with previously conducted trials of golimumab.4 5 16 Finally, more patients met the EE criteria in the golimumab 50 mg + MTX group (Group 2) than in the golimumab 100 mg + MTX group (Group 3), indicating the potential for a dose response.
In interpreting the efficacy findings of this study, it is important to bear in mind that patients could enter this study based on measures of disease activity generally considered to be subjective in nature (ie, tender and swollen joint counts and morning stiffness) or reported from each trial site (ESR) without confirmation by centrally determined parameters such as CRP or erosions. This could have resulted in study enrolment of patients with relatively inactive disease.
Golimumab was generally well tolerated with no unexpected safety issues observed in Japanese patients with RA. By week 24, approximately 10% of all patients treated with golimumab + MTX had an injection site reaction. A variety of dermatological adverse effects, including injection site reactions and dermatitis, have been reported for TNF antagonists such as adalimumab, etanercept and infliximab,20 as well as for anakinra, a recombinant human form of interleukin-1 receptor antagonist.21 These dermatological complications typically are well-tolerated, respond to antihistamines and do not necessitate treatment discontinuation.
The incidences of serious AEs, serious infections and malignancies during the fixed treatment regimen period were low and similar with placebo + MTX (1.1%, 0.0% and 0.0%, respectively) and combined golimumab + MTX (1.7%, 0.6% and 0.0%, respectively). These findings indicate a safety profile similar to placebo + MTX (2.3%, 0.8% and 0.0%, respectively) and golimumab + MTX (7.3%, 3.9% and 1.1%, respectively) at week 16 in the GO-FORWARD study.5 However, these safety findings must be interpreted with caution given the relatively small number of patients evaluated, the lack of power to detect treatment group differences in individual safety events and the relatively short follow-up period. No patients died and no cases of tuberculosis were documented during the 24-week study period.
Taken together, the efficacy and safety findings presented here indicate that golimumab 50 mg + MTX and golimumab 100 mg + MTX were at least as safe and effective in these Japanese patients with active RA despite MTX therapy as they were observed to be when administered to patients with RA who also had an inadequate response to MTX in the GO-FORWARD study.5
Acknowledgments
The authors thank the patients, investigators and study personnel who made this trial possible, and Kirsten Schuck, Michelle Perate MS and Mary Whitman PhD (Janssen Services, LLC) for their assistance in preparing, editing and submitting the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Adobe PDF - ARD_text_317.pdf
Footnotes
-
Funding This study was funded by Centocor Research & Development Inc, Janssen Pharmaceuticals KK and Mitsubishi Tanabe Pharmaceutical Corporation.
-
Competing interests DB is an employee of Centocor, a fully-owned subsidiary of Johnson & Johnson and owns stock in Johnson & Johnson. HY has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical, Otsuka Pharmaceutical, Roche, Takeda Pharmaceutical and Wyeth. KY has received research grants from Astellas Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical, Immunofuture Inc, Mitsubishi Tanabe Pharmaceutical, Santen Pharmaceutical and Wyeth. MH has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical, Takeda Pharmaceutical and Wyeth, as well as consulting fees from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Janssen Pharmaceutical and Mitsubishi Tanabe Pharmaceutical. MK has received research grants from Astellas Pharmaceutical, Astra Zeneca, Banyu Pharmaceutical, Daiichi Sankyo Pharmaceutical, Eizai Pharmaceutical, GlaxoSmith Kline, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical and Nippon Boehringer Ingelheim. NI has received research grants from Astellas Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical and Mitsubishi Tanabe Pharmaceutical. NM has received research grants from Abbott, Astellas Pharmaceutical, Banyu Pharmaceutical, Chugai Pharmaceutical, Daiichi Sankyo Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical, Takeda Pharmaceutical and Teijin Pharmaceutical. TK has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical, Otsuka Pharmaceutical, Pfizer, Takeda Pharmaceutical and Wyeth. TO is an employee of Janssen Pharmaceutical KK, a fully-owned subsidiary of Johnson & Johnson. TT has received research grants from Abbott, Astra Zeneca, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical, Novartis, Takeda Pharmaceutical and Wyeth. TY is an employee of Mitsubishi Tanabe Pharmaceutical. YT has received research grants from Abbott, Astellas Pharmaceutical, Banyu Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical, Pfizer and Takeda Pharmaceutical.
-
Provenance and peer review Not commissioned; externally peer reviewed.