Article Text

Statistics from Altmetric.com
Rheumatoid arthritis (RA) is a chronic inflammatory disease that causes progressive joint destruction and consequently functional disability due to the combined effect of chronic synovitis and progressive joint damage. Treatment with disease-modifying drugs and biological agents improves pain, fatigue and disability. More recently, the concept of tight control has been introduced in the treatment of RA. Tight control may be defined as a treatment strategy tailored to the disease activity of individual patients, with the aim of achieving a predefined level of low disease activity or preferably remission within a reasonable period of time.1 A goal of remission is reasonably to halt joint damage. However, patients with RA in remission by any established criteria can still experience radiographic progression.2 In current clinical practice, as well as in most clinical trials, joint destruction is not the explicit target for treatment. Also more recent strict criteria for remission do not take actual joint damage into account and focus on disease activity.3 In contrast, intensive therapy with antitumour necrosis factor (TNF) alpha plus methotrexate results in no or minimal radiographic progression irrespective of patients' disease activity.4 This dissociates clinical disease activity from the progression of joint damage. If the prevention of joint damage is a major goal of treatment of RA, monitoring the progression of joint damage more closely is reasonable and needs more attention in future studies.5 Of course, this should be in addition to controlling disease activity as an expression of, predominantly, joint inflammation. It can be speculated that disease with dominating joint damage and disease with dominating inflammation might express the boundaries of different phenotypes of RA.
Evidently, (autoimmune) inflammation as seen in RA constitutes a crucial aspect in disease activity. Investigations concentrate largely on these immune processes. A complex network of immune cells is in charge of an even more complex network of inflammatory cytokines and chemokines that form the root of disease activity. Although less in the spotlight, concurrent with this cytokine activity, a more specialised local complex of activities between local synovial cells, particularly fibroblasts, and the neighbouring cartilage and bone tissue, is active at the margins of the joint. As this activity causes actual joint damage, this is also presumed to be a critical process in RA. This local pathology may at least partly be independent of synovial inflammation as a general phenomenon. The pathogenesis may indirectly respond to disease activity-directed strategies, but clearly it is not the direct target for anti-inflammatory treatment. In contrast, local synovial fibroblast activity and cartilage damage may enhance general inflammation (disease activity) in RA. It is anticipated that this local synovial activity can act autonomously and mediate cartilage and bone destruction, more or less independently of general synovial inflammation. The success of therapeutic intervention targeting inflammatory cytokines and cell interactions in RA has underlined the importance of the respective key players such as T cells, B cells and macrophages, with their soluble mediators such as TNF, interleukin (IL)-6, and many others. Clearly, they are not the only players as there is no unambiguous coupling between disease activity representing inflammation and the severity of disease representing actual joint tissue destruction.6
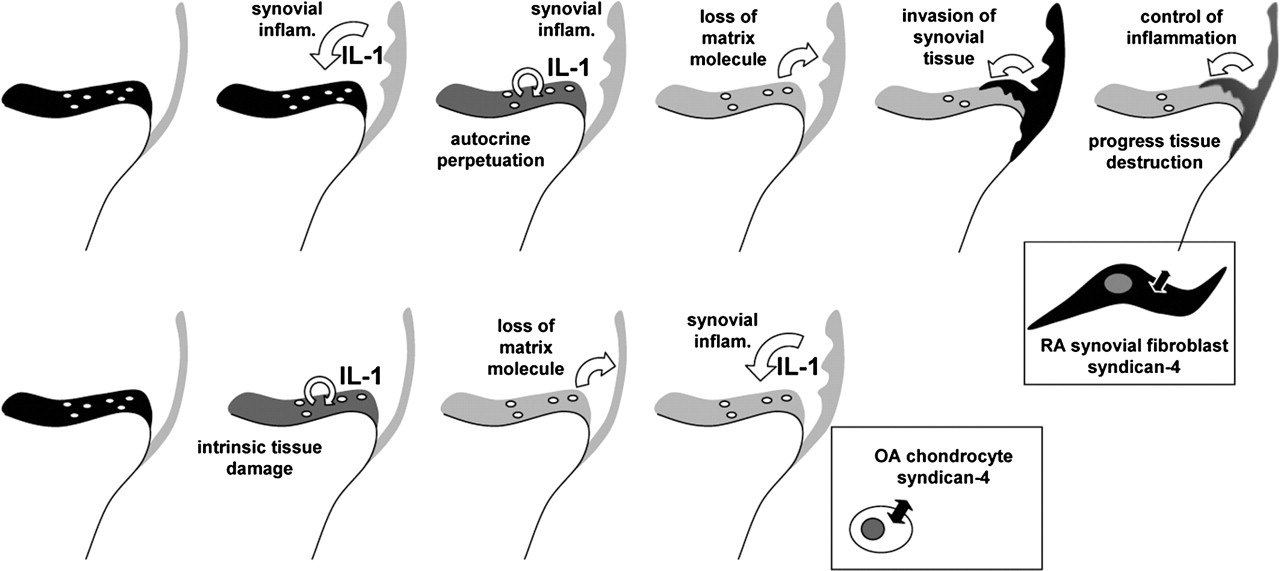
In this issue of Annals of Rheumatic Diseases a study is described that provides potential mechanisms for the progression of joint damage despite control of inflammation. This may lead to potential targets for additional (co-)therapy. The study of Korb and colleagues from the rheumatology departments of Vienna Medical University in collaboration with Munster and Erlangen in Germany is entitled: ‘Early structural changes and bone are required for pannus attachment and invasion during destructive inflammatory arthritis’.7 The authors used a human TNF transgenic mouse model of spontaneous developing destructive arthritis mimicking human RA with cartilage and bone destruction. The study reports that general synovial inflammation caused by RA results in cartilage matrix destruction by the loss of proteoglycans from the cartilage matrix. Subsequently, this cartilage damage triggers synovial tissue attachment and invasion into the cartilage matrix (figure 1). Loss of cartilage matrix in the initial phase is not dependent on contact between synovial tissue and cartilage. As such, cartilage damage precedes synovial fibroblast invasion. This activity appears to be dependent on IL-1. IL-1, among others, plays a role in this early cartilage damage, as they suggest, and may also be ‘transforming’ the synovial fibroblast to turn into an aggressive phenotype. Collagenase can also induce cartilage damage and enable synovial fibroblasts to invade the cartilage, although less profoundly than IL-1. Normal (non-RA) fibroblasts hardly attach to cartilage, also not after IL-1 pretreatment. As such, the destructive properties of the fibroblast appear to be guided by damaged cartilage but are also intrinsic to the RA phenotype of the fibroblasts. Syndycan-4 deficiency (knockout mice) limits invasion (adhesion) of fibroblasts into (on) cartilage. It remains unclear from the described study whether this is due to deficiency in synovial fibroblast syndycan-4 or whether syndycan-4 deficiency in chondrocytes is involved. The deficiency of IL-1 (knockout mice) does not affect the inflammatory aspects of synovitis, but prevents cartilage degradation and reduces bony destruction. Importantly, these results provide a biological/molecular mechanism of the clinically observed uncoupling of inflammation and tissue destruction; a concept that was postulated decades ago.8
{kind=link}
Top row from left to right: Sequence of events in rheumatoid arthritis (RA); left in black represents cartilage with chondrocytes on top of bone, with synovial tissue lining. Synovial inflammation triggers cartilage damage and loss of cartilage matrix components. An intrinsic process in cartilage is anticipated to perpetuate this process, as chondrocytes themselves can produce significant amounts of IL-1. The ‘sensitised’ matrix triggers the synovial fibroblast to turn into an aggressive phenotype invading the cartilage. Right: Control of inflammation (disease activity-targeted treatment) will control inflammation and with that indirectly in part synovial tissue invasion and subsequently indirectly in part cartilage and bone erosion. The uncontrollable part may depend on an autonomic process not directly controlled by inflammation. Syndican-4 expression appears crucial in fibroblast adhesion and invasion. Co-treatment targeting this synovial tissue invasion (pannus formation) might add to stop joint destruction in RA, maybe particularly in certain specific phenotypes of the disease. Bottom row from left to right: The process of joint damage in osteoarthritis (OA). Expectedly, a mechanically triggered and subsequently intrinsic cartilage destructive process, in which IL-1 plays an essential role, leading to secondary inflammation, which adds to further cartilage damage, among others by IL-1, in which the chondrocyte syndican-4 plays a roll in matrix degradation, but in which synovial tissue hardly ever becomes invasive and tissue aggressive as observed in RA.
The question arises, what makes these synovial fibroblasts become aggressive, tissue invasive cells? In the study referred to,7 IL-1 and syndycan-4 are suggested to play crucial roles. Syndycan-4 is even suggested to constitute a novel target for the treatment of arthritic cartilage damage. Is that indeed the way to proceed?
Interestingly, IL-1 and syndycan-4 are also key players in osteoarthritis, whereas osteoarthritis is hardly ever affected by synovial tissue invasion, designated as pannus formation (figure 1). Osteoarthritis is mostly a degenerative disease with intrinsic cartilage degeneration and only secondary inflammation. More recently, osteoarthritis synovial inflammation has become of increasing importance, and even the focus of treatment approaches in osteoarthritis. Inflammation dominated, more erosive forms of osteoarthritis, might represent a specific phenotype, which needs specifically more aggressive anti-inflammatory treatment.9 Even in these cases reports on aggressive invading and overgrowing synovial tissue fibroblasts are scares if not absent.
IL-1 plays a dominant role in osteoarthritis cartilage degeneration, in which osteoarthritis chondrocytes produce significant amounts of this cytokine, driving the destructive process. Subsequent synovial inflammation adds to IL-1 activity.10 This may question whether IL-1 can be designated as the key component in changing the synovial fibroblast into an aggressive phenotype, because osteoarthritis is not specifically characterised by this phenomenon. In the in-vivo TNF mouse model the absence of IL-1 prevented clear adhesion of synovial fibroblast to cartilage,7 but collagenase was also able to change cartilage properties in such a way that arthritic fibroblasts could adhere. As the authors mention, Ishikawa and colleagues11 in 1991 reported the importance of IL-1, but not uniquely IL-1, in synovial invasion. IL-1 enhanced the process by releasing endogenous proteolytic enzymes from chondrocytes. Increased binding following collagenase pretreatment of cartilage was observed in these experiments, and interferon was unable to induce these effects. Compared with TNFα, blocking IL-1 shows particularly strong protective effects on cartilage and bone erosion in all of the mouse models including antigen and non-antigen-induced arthritis.12 13 A more recent study using human cartilage and RA synovial fibroblasts transplanted into severe combined immunodeficient mice demonstrated that IL-1 stimulates articular chondrocytes to produce factors that act on fibroblast, increasing their destructive potential. In these studies direct effects of IL-1 on fibroblast activity might also have been involved. Interestingly, the addition of human TNFα did not enhance the degradation of the cartilage-like matrix.14 In contrast, synovial fibroblasts transfected with IL-R antagonist appear still able to attach and invade into cartilage.15 As such it may be concluded that IL-1 is a key player in cartilage damage in RA and osteoarthritis. For osteoarthritis this is supported by the effect of the inhibition of IL-1 in animal models16 and for RA by the effect of anakinra, an IL-1 receptor antagonist, in diminishing radiographic joint damage.17
The direct effects of IL-1 on synovial fibroblasts may be stimulatory, but are not the key component; otherwise osteoarthritis would also be characterised by synovial tissue invasion, which is not the case. Apparently, it needs more to change synovial fibroblast to become aggressive than just IL-1 and IL-1-driven cartilage damage. When synovial fibroblasts isolated from RA patients are implanted with human cartilage into severe combined immunodeficient mice, synovial fibroblasts invaded cartilage and showed destructive behaviour in the absence of other immune cells, suggesting that this is an intrinsic characteristic of the RA phenotype of these synovial fibroblasts. This explains the difference between the aggressive behaviour in RA and the absence of such behaviour in osteoarthritis. In both cases cartilage is deprived of matrix components by the activity of IL-1, and IL-1 can act on the fibroblast present. Irrespective of this, in osteoarthritis the synovial fibroblasts do not become as aggressive.18 19
Based on the demonstration that chondrocytes can produce proinflammatory cytokines such as IL-1, it has been hypothesised for some time that chondrocytes may contribute to the pathogenesis not only of osteoarthritis but also RA by various cell/matrix components.20 The authors propose syndycan-4 as a key player,7 but clearly more mediators have been proposed as such. As an example, extracts of cartilage from patients with RA as well as osteoarthritis contain a range of fibronectin fragments. These fibronectin fragments are both autocrine and paracrine regulators of cartilage damage.19 Increased levels of these fragments, found in osteoarthritis and RA joints stimulate cartilage breakdown.22 Such cartilage-associated molecules, like fibronectin, enhance the aggressive growth of RA synovial fibroblasts.21 Apart from fibronectin (fragments) syndecan-4 from cartilage stimulate synovial fibroblast ingrowth.21 22 The absence of syndycan-4 (knockout) diminishes arthritic synovial fibroblast adhesion significantly, as demonstrated in the study of Korb et al.7 Syndecan-4 is also upregulated in osteoarthritis and the inhibition of syndecan-4 reduces cartilage destruction in, for example, a mouse models of osteoarthritis.23 This has suggested the inhibition of syndecan-4 to be a specific target for the treatment of cartilage damage also in osteoarthritis. Cartilage-associated molecules, such as fibronectin and syndican-4, enhance the aggressive growth of the RA synovium.24 Apparently, as also significantly expressed in osteoarthritis, these molecules are not the key component; otherwise osteoarthritis would also be characterised by synovial tissue invasion, which is not the case. Korb and colleagues7 have, to our opinion, still not unfolded the mystery of how synovial fibroblasts ‘transform’ into aggressive, invasive cells, but they do identify an intriguing mechanism of how invasion into cartilage is facilitated in vivo.
The study contributes to the accumulating evidence that cartilage is not just a passive tissue that undergoes destruction by synovial inflammation. Rather, primary cartilage damage appears to constitute an important prerequisite in the joint pathogenesis of RA. The aggressive synovial tissue activity might become (at least partly) an independent process of the general inflammation. As such, co-medication targeted at the inhibition of destruction in addition to inhibitors of inflammation might be warranted. This could address the so far underrepresented aspects of RA pathogenesis such as the switch from acute to chronic inflammation and the early and perpetual loss of cartilage and bone.
Whether interfering with syndican-4 cell attachment constitutes an effective approach for treating cartilage damage in RA remains to be established. However, the fact that new pathways of tissue destruction in RA, such as that described, need to be explored to get a full grip on the disease activity as well as severity is indisputable.
References
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.