Article Text

Abstract
Objectives To investigate the expression and activation of mitogen-activated protein kinases in patients with early arthritis who are disease-modifying antirheumatic drug (DMARD) naïve.
Methods A total of 50 patients with early arthritis who were DMARD naïve (disease duration <1 year) were prospectively followed and diagnosed at baseline and after 2 years for undifferentiated arthritis (UA), rheumatoid arthritis (RA) (1987 American College of Rheumatology (ACR) and 2010 ACR/European League Against Rheumatism (EULAR) criteria), or spondyloarthritis (SpA). Synovial biopsies obtained at baseline were examined for expression and phosphorylation of p38, extracellular signal regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) by immunohistochemistry and digital analysis. Synovial tissue mRNA expression was measured by quantitative PCR (qPCR).
Results ERK and JNK activation was enhanced at inclusion in patients meeting RA criteria compared to other diagnoses. JNK activation was enhanced in patients diagnosed as having UA at baseline who eventually fulfilled 1987 ACR RA criteria compared to those who remained UA, and in patients with RA fulfilling 2010 ACR/EULAR criteria at baseline. ERK and JNK activation was enhanced in patients with RA developing progressive joint destruction. JNK activation in UA predicted 1987 ACR RA classification criteria fulfilment (R2=0.59, p=0.02) after follow-up, and disease progression in early arthritis (R2=0.16, p<0.05). Enhanced JNK activation in patients with persistent disease was associated with altered synovial expression of extracellular matrix components and CD44.
Conclusions JNK activation is elevated in RA before 1987 ACR RA classification criteria are met and predicts development of erosive disease in early arthritis, suggesting JNK may represent an attractive target in treating RA early in the disease process.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Mitogen-activated protein kinase (MAPK) family members, namely p38 kinases (α–δ), extracellular signal regulated kinases (ERKs) 1 and 2, and c-Jun N-terminal kinase (JNKs) 1–3, occupy critical positions in coupling diverse cell surface proteins, including antigen receptors, tumour necrosis factor (TNF) family receptors, chemokine and cytokine receptors, and Toll-like receptors to inflammatory gene expression.1 2 Members of each MAPK family are expressed and activated in synovial tissue of patients with rheumatoid arthritis (RA) and other forms of inflammatory arthritis.3,–,5 Highly selective pharmacological inhibitors of p38,6,–,9 ERK10 11 and JNK3 12 13 prevent inflammatory activation of stromal fibroblast-like synoviocytes (FLS) derived from synovial tissue, chondrocytes and osteoclasts from patients with RA. Additionally, pharmacological inhibition or genetic deletion of MAPK activity reduces inflammation and joint destruction in multiple experimental animal models of RA.6 8 10 12 14,–,18 These data collectively suggest that therapeutic strategies inhibiting MAPK activation may be useful in the treatment of RA.1 2 19 20
Despite this wealth of preclinical analyses, little is known about the distinct contributions of each MAPK to the onset and perpetuation of RA. Clinical parameters and biomarkers have yet to be identified which are associated with synovial MAPK activation status, and MAPK activation in RA has primarily been examined in patients with destructive end-stage disease.3,–,5 In the transgenic human TNF model of murine arthritis, p38 activation is required for induction of inflammation and joint destruction.15 21 Whether this observation can be translated into successful treatment of RA with MAPK inhibitors, especially in early disease, is uncertain however, as clinical trials with p38 inhibitors have not been successful.22 23 Recent kinetic analyses of MAPK activation in experimental murine arthritis have revealed model-specific differences in the degree of p38, ERK and JNK activation, as well as in the timing of their activation during disease onset and resolution.24 Here, we examined if similar differences in MAPK involvement might be relevant to the earliest stages of the development of RA, by assessing the relationship between MAPK expression and activation, and disease diagnosis and outcome in a prospective cohort of patients with early arthritis who were disease-modifying antirheumatic drug (DMARD) naïve.
Patients and methods
Patients
A total of 50 patients with arthritis of duration of less than 1 year, as measured from the first clinical signs of arthritis, irrespective of which joint was initially affected, and a clinically inflamed knee, ankle or wrist joint, underwent arthroscopic synovial biopsy. Diagnosis of RA or spondyloarthritis (SpA) was made according to established classification criteria.25,–,28 Patients were classified as having undifferentiated arthritis (UA) if no classifying diagnosis for RA, SpA or other forms of arthritis could be made. After 2 years of follow-up final diagnosis was made according to classification criteria. All patients were naïve to treatment with DMARDs and corticosteroids at inclusion, and after baseline study procedures all patients were treated consistent with European League Against Rheumatism (EULAR) guidelines.29 In case of a clinical diagnosis of RA, DMARD treatment was initiated directly after baseline study procedures were completed. The 28-joint Disease Activity Score (DAS28) was systematically determined and patients were treated according to the treat-to-target principle, aiming for DAS28 <2.6, using conventional DMARDs, corticosteroids and biologicals, if indicated. If a combination of DMARDs did not result in a DAS28 <3.2 then a biological was given. Upon decision of the treating doctor corticosteroids were given in combination with a DMARD, either high dose and tapered down in 6–8 weeks or more prolonged low dose. In case of a diagnosis of SpA with peripheral arthritis, all patients, except patients with reactive arthritis were started on methotrexate. The patients with reactive arthritis were treated with intra-articular steroids and non-steroidal anti-inflammatory drugs. Treatment was aimed at minimal disease activity. Patients with UA were treated with intra-articular steroids and, if arthritis was persistent, a DMARD was given.
At inclusion we assessed disease activity (68 tender and 66 swollen joint score, patient's visual analogue scale (VAS) of global disease activity (scale 0–100 mm), VAS of pain (scale 0–100 mm), erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels) and collected serum and arthroscopic synovial tissue biopsy samples. X-rays were obtained at baseline and after 2 years; erosion scoring was based on the presence or absence of erosions on x-rays of hands and feet where the modified Sharp-van der Heijde erosion score was ≥1.30 This study was approved by the institutional review board, performed according to the declaration of Helsinki. All study patients provided written informed consent.
Measurement of serum autoantibodies
Patient serum immunoglobulin M rheumatoid factor (IgM-RF) and anti-citrullinated protein antibodies (ACPA) were measured using IgM-RF (Sanquin, Amsterdam, The Netherlands) and anti-cyclic citrullinated peptide 2 (CCP2) (Eurodiagnostica, Arnhem, The Netherlands) ELISA kits.
Synovial tissue biopsy sampling, immunohistochemistry and gene expression
All patients underwent arthroscopic synovial tissue biopsy sampling of an actively inflamed ankle, wrist or knee joint.31 Synovial tissue biopsies were collected from each patient, stored and processed for immunohistochemistry, and stained sections analysed by digital imaging analysis as previously described.32,–,34 Detailed descriptions of stainings, analyses and mRNA expression studies are provided in the supplementary methods.
Statistics
Statistical analysis was performed using Windows GraphPad Prism 4 (GraphPad Software, La Jolla, California, USA) and SPSS V.16.0 (SPSS, Chicago, Illinois, USA) software. Comparisons in expression or phosphorylation of markers between cohorts were performed using the Mann–Whitney U test, first using the Kruskal–Wallis test when more than two groups were compared. Logistic regression analysis was used to analyse the relationship between relative MAPK phosphorylation and development of RA and the development of erosive disease. Results were considered statistically significant if p<0.05.
Results
p38, ERK and JNK are differentially expressed and phosphorylated in patients with early arthritis with distinct diagnoses
We performed immunohistochemical staining on synovial biopsy samples using antibodies recognising total and p-p38, ERK and JNK MAPKs. Within the cohort, 27 patients were diagnosed as having RA at 2 years after enrolment in the study based on 1987 American College of Rheumatology (ACR) criteria for RA, 7 with SpA and 16 with UA. Clinical characteristics of each patient group are shown in table 1. p38 expression was highest in UA, and significantly different in this group compared to SpA (p<0.05), but not RA (supplementary figure 1). The p-p38 levels were higher in UA than SpA (p<0.05). ERK expression was significantly higher in RA than SpA (p<0.01) and UA (p<0.01) (supplementary figure 1). p-ERK levels were also highest in RA, where it was elevated compared to SpA (p<0.01) and UA (p<0.01). No differences in JNK expression were noted between diagnostic groups (supplementary figure 1), but p-JNK levels were higher in RA than in SpA (p<0.005) and UA (p<0.01). Within each diagnostic group, and across the cohort as a whole, no significant correlation was observed between MAPK expression and phosphorylation levels, suggesting that phosphorylation was driven by differential inflammatory stimuli input (data not shown, see supplementary figure 2 for representative photomicrographs of staining distribution in each diagnostic group).
Characteristics of study patients
Relative phosphorylation of ERK and JNK is enhanced in patients with early RA
To gain insight into the degree of MAPK engagement by inflammatory stimuli, we calculated the relative phosphorylation of p38, ERK and JNK proteins for each patient (IOD phospho-MAPK/IOD total MAPK, arbitrary units). Relative p38 phosphorylation was similar in all diagnostic groups (figure 1A, left panel). In contrast, relative ERK phosphorylation in RA was higher than in SpA (p<0.05) and UA (p<0.01) (figure 1A, middle panel). Relative JNK phosphorylation (figure 1A, right panel) was elevated in RA compared to UA (p<0.01) and SpA (p<0.005). Using 2010 ACR/EULAR classification criteria, 35 patients were diagnosed as having RA, 7 with SpA and 8 with UA at baseline (supplementary table 1). Here, no differences in relative p38 or ERK phosphorylation were observed, but relative JNK phosphorylation was elevated in RA (p<0.001) compared to SpA (figure 1B).

Relative mitogen-activated protein kinase (MAPK) activation in relation to disease diagnosis. Quantitative comparison of relative MAPK phosphorylation in the synovial tissue of patients with early arthritis. Tissue sections from patients diagnosed as having rheumatoid arthritis (RA) according to 1987 American College of Rheumatology (ACR) criteria, spondyloarthritis (SpA) and undifferentiated arthritis (UA) after 2 years of follow-up were stained with antibodies against phospho- and total p38, extracellular-signal regulated kinase (ERK) and c-Jun N-terminal kinase (JNK). Stainings were developed with biotin tyramide enhancement, horseradish peroxidase and aminoethylcarbazole, followed by counterstaining with Mayer's haematoxylin, and evaluated by digital imaging analysis. Relative phosphorylation levels (ratio of IOD phosphorylated protein to IOD of total protein, arbitrary units) of p38, ERK and JNK were calculated for patients diagnosed as having RA according to (A) 1987 ACR criteria and (B) 2010 ACR/European League Against Rheumatism (EULAR) criteria, SpA and UA. Data are presented as boxplots, where the boxes represent the 25th to 75th percentiles, the lines within the box mark the median value, and lines outside the boxes denote the 10th and 90th percentiles. Lines connecting data sets indicate statistically significant differences between groups. *p<0.05; **p<0.01; ***p<0.005.
Relative phosphorylation of ERK and JNK is enhanced in patients with early RA before fulfilment of 1987 ACR RA classification criteria
We next compared MAPK activation with the presence of RF and ACPA, autoantibodies predictive of the development, disease course and prognosis of RA.35 36 Patients with RA and UA receiving a diagnosis of RA at 2 year follow-up (UA>>RA) identified by 1987 ACR criteria were grouped based on the absence or presence of serum RF or ACPA at baseline (see supplementary tables 2 and 3 for patient clinical characteristics). Relative activation of ERK (p<0.0005) and JNK (p<0.005) was significantly higher in patients who were RF+ (n=14) than RF– (n=13). Patients who were ACPA+ (n=13) also displayed enhanced ERK (p<0.001) and JNK (p<0.005) activation compared to patients who were ACPA– (n=14) (supplementary figure 3).
Characteristics of patients with rheumatoid arthritis (RA) with erosive and non-erosive disease
Characteristics of patients with early arthritis with erosive and non-erosive disease
Given that ERK and JNK activation was elevated in patients who were RF and ACPA seropositive, we next examined if ERK and JNK might already be activated in UA>>RA, as diagnosed by 1987 ACR criteria (see table 1 for patient characteristics). We observed no differences in relative p38 (figure 2A, left panel) or ERK (figure 2A, middle panel) phosphorylation between patients diagnosed as having UA who remained UA (UA>>UA) (n=16) and patients who progressed from UA>>RA (n=8). In contrast, JNK phosphorylation was elevated in UA>>RA (figure 2A, right panel) compared to UA>>UA (p<0.005). Univariate logistic regression analysis showed that relative JNK activation at baseline was significantly related to fulfilment of classification criteria for RA after follow-up with an explained variance of 59% (R2=0.59, p=0.02). Thus, in patients with early arthritis, elevated ERK and JNK activity distinguish RA from other forms of arthritis, and JNK activation is elevated in patients with RA even before 1987 ACR classification criteria of RA are met. Similar regression analyses based on 2010 ACR/EULAR classification criteria could not be performed due to the small number (n=2) of patients with UA>>UA (supplementary figure 4).

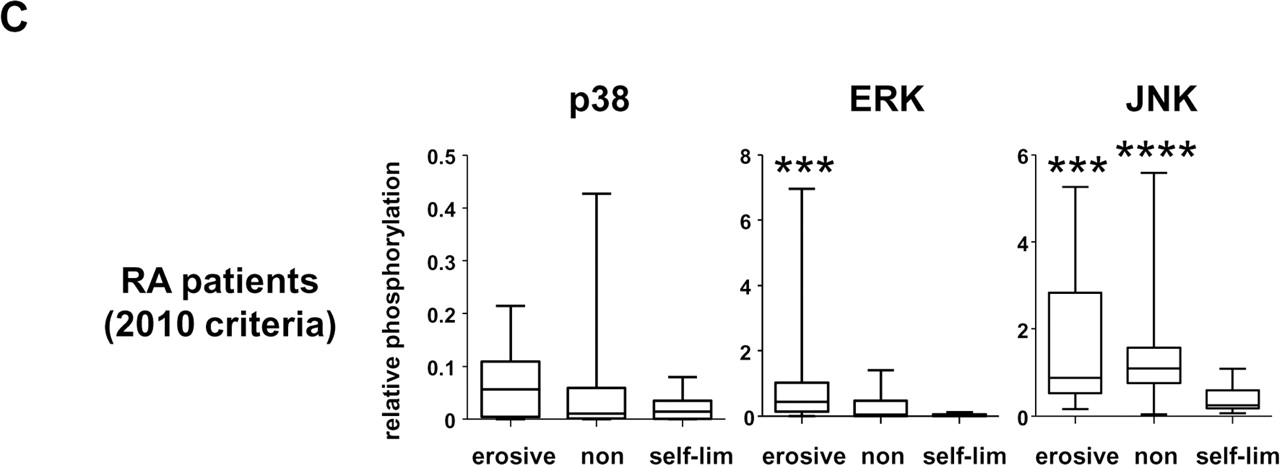
Relative mitogen-activated protein kinase (MAPK) activation in relation to disease outcome and progression. A. Quantitative comparison of relative MAPK phosphorylation in the synovial tissue of patients classified according to 1987 American College of Rheumatology (ACR) criteria as undifferentiated arthritis (UA) which remained UA after 2 years (UA>>UA), and UA which was diagnosed as rheumatoid arthritis (RA) after 2 years (UA>>RA). Relative phosphorylation levels of p38, extracellular signal regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) were calculated for patients diagnosed as having UA>>UA and UA>>RA. B. Quantitative comparison of relative MAPK phosphorylation in the synovial tissue of patients classified as having RA after 2 years based on 1987 ACR criteria (upper panels) and all patients with early arthritis with persistent erosive and persistent non-erosive disease (lower panels). C. Comparison of relative MAPK phosphorylation in the synovial tissue of patients classified with RA according to 2010 ACR/EULAR criteria displaying self-limiting, persistent non-erosive and erosive disease. Erosive disease was defined by a Sharp-van der Heijde erosion score ≥1 at 2 year follow-up. Relative MAPK phosphorylation was calculated as the ratio of IOD phosphorylated protein to IOD of total protein (arbitrary units). Data are presented as boxplots, where the boxes represent the 25th to 75th percentiles, the lines within the box mark the median value, and lines outside the boxes denote the 10th and 90th percentiles. Lines connecting data sets indicate statistically significant differences between groups. *p<0.05; **p<0.01; ***p<0.005.
Relative ERK and JNK activation is elevated in patients with RA developing erosive disease
To examine the relationship between MAPK activation and disease outcome in patients with RA, RA>>RA and UA>>RA were pooled and grouped based on the development of erosive disease (defined as a modified Sharp-van der Heijde erosion score was ≥1 at 2 year follow-up, see table 2 for patient clinical characteristics).30 Only one patient with early arthritis had erosive disease at baseline. Relative p38 phosphorylation was similar between patients with RA with non-erosive (n=15) and erosive disease (n=12) (figure 2B, upper left panel). Relative ERK (p<0.01) (figure 2B, upper middle panel) and JNK phosphorylation (p<0.01) (figure 2B, upper right panel) was higher in patients developing erosive disease. Examining all patients with early arthritis, regardless of diagnosis (see table 3 for patient characteristics), activation of p38 (p<0.05), ERK (p<0.005) and JNK (p<0.001) was elevated in patients with erosive disease (figure 2B, lower panels). JNK activation, but not p38 or ERK activation, predicted development of erosive disease (R2=0.16, p<0.05).
Relative JNK activation and is enhanced in patients with persistent disease
Among patients who fulfilled 2010 ACR/EULAR criteria for RA after 2 years of follow-up, 17 patients had persistent non-erosive disease, 12 had erosive disease and 10 had self-limiting disease (supplementary table 4). ERK activation (figure 2C) was elevated in patients with erosive disease compared to those with self-limiting disease (p<0.005), and a trend towards enhanced ERK activation was observed in patients with erosive disease compared to those with persistent non-erosive disease (p=0.051). JNK activation was significantly lower in those patients with RA with self-limiting disease compared to those with persistent (p<0.005) and erosive disease (p<0.01), associated with lower scores for tender joint count, swollen joint count and DAS28 (supplementary table 4).
As JNK activation was elevated in persistent disease, and often associated with the regulation of expression extracellular matrix (ECM) component biology and cellular interactions with the ECM, we examined expression of a targeted gene set in patients with RA from this cohort with self-limiting (n=8) and persistent (non-erosive and erosive, n=14) disease from which mRNA was available. Quantitative PCR (qPCR) analysis was performed on approximately 80 genes encoding ECM components, matrix metalloproteinases (MMPs) and adhesion molecules. Unsupervised hierarchical cluster analysis of gene products failed to group patients according to disease outcome (data not shown), and no trends toward similar gene expression patterns were observed using supervised hierarchical clustering (figure 3A). However, analysis of expression of each individual gene (figure 3B and supplementary table 5) revealed that expression of collagens COL1A1, COL4A2, COL5A1, COL6A1 and COL16A1, the laminin LAMC1 and MMP-12 were significantly downregulated in patients with persistent disease, while expression of CD44 was elevated (all p<0.05).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gene expression analysis in synovial tissue of patients with early rheumatoid arthritis (RA) with self-limiting and persistent disease. A. Supervised hierarchical cluster analysis of expression of 83 genes involved in extracellular matrix (ECM) biology and cellular adhesion. Analysis was performed on patients with RA, undifferentiated arthritis (UA)>>UA and UA>>RA, pooled and grouped based on self-limiting (n=8) and persistent (n=14) disease. Each column represents the data of one patient and each row shows the relative expression of a given gene for all patients. Gene designations are listed at the right of the figure. Red signifies relatively higher expression of a given gene, green relatively lower expression and black signifies equivalence to the median expression of that gene across all patients. B. Relative mRNA expression of selected genes in patients with self-limiting and persistent disease, expressed as the ratio between the gene of interest and ribosomal protein L13a (RPL13A). Each data point represents a value for an individual patient and bars represent mean values. *p<0.05.
Discussion
Because p38, ERK and JNK are each detected in their activated form in the synovial tissue of patients with various forms of inflammatory arthritis, and are requisite for pathology in animal models of arthritis, MAPKs might represent attractive therapeutic targets in the treatment of RA and other forms of arthritis.1,–,4 However, the clinical inefficacy of p38 inhibitors in early trials with patients with RA has indicated a need for a greater understanding of the contributions of MAPKs to synovitis and joint destruction.22 23 37 Intriguingly, recent kinetic analyses of MAPK activation in multiple murine models of RA have also indicated model-specific differential involvement of each MAPK in distinct phases of initiation, perpetuation and resolution of disease.24 This latter observation prompted us to examine MAPK expression and phosphorylation in the earliest stages of human arthritis.
We find that synovial activation of ERK and JNK, but not p38, is significantly elevated in patients with RA compared to patients with other diagnoses, as well as in patients who develop erosive RA. Perhaps important to the aetiology of RA, JNK activation is elevated in patients with RA even before 1987 ACR classification criteria of RA are met. Assessing all patients with early arthritis, regardless of disease diagnosis, we found that each MAPK was more highly activated in patients with erosive disease, but here, only JNK activation predicted the development of erosive disease. Forward and backward linear regression failed to identify roles for p38 and ERK in diagnosis and disease progression (data not shown). Further studies in larger independent cohorts will obviously be needed to assess the positive predictive value and the negative predictive value of JNK activation in predicting RA and erosive disease.
We found no evidence supporting a role for p38 in the onset of RA or eventual joint destruction in arthritis. Phosphorylated p38 is readily detected in the synovial tissue of patients with active and end-stage destructive RA, and mice in the human TNF-transgenic model of RA.3 4 21 In these mice, pharmacological p38 inhibition blocks disease onset.15 However, p38 activation in murine collagen-induced arthritis is only modestly increased over baseline until late in disease (day 40–50), when clinical parameters and cytokine biomarkers of disease activity recede.24 Also, p38 activity is not affected in TNF-transgenic mice following anti-TNF treatment, or in patients with psoriatic arthritis treated with etanercept.21 38 Thus, p38 may contribute significantly to negative feedback mechanisms dampening inflammatory responses or repairing tissue in established RA.39 In patients with early arthritis p38-dependent repair mechanisms may not yet have been initiated.
ERK phosphorylation is also elevated in patients with early arthritis diagnosed as having RA and in patients with RA who develop erosive disease. ERK phosphorylation is also readily detected in the synovial tissue of patients with RA undergoing joint replacement.4 The extent and timing of ERK activation in experimental arthritis is heavily model-dependent, although genetic or pharmacological suppression of ERK appears to uniformly suppress disease.10 17 24 However, as in vitro studies in RA FLS have revealed that many secreted products routinely assessed as TNF-dependent activation markers relevant to RA are relatively insensitive to ERK inhibition. Thus, the mechanisms by which ERK contributes to inflammation and disease progression in the earliest stages of RA remain to be established.9
Strikingly, JNK phosphorylation is significantly elevated in synovial tissue from patients with early RA, even before 1987 ACR classification criteria are met. During the course of our study, new classification criteria for the diagnosis of RA were defined,27 28 and re-examining our cohort with these criteria, we found that half of the patients originally classified as having UA at baseline now met criteria for RA, similar to a recent study.40 Importantly, using the new criteria, we confirmed that relative JNK activity is enhanced in early RA. However, insufficient numbers of patients could be classified as UA>>UA to allow logistic regression analysis to determine whether JNK activity at baseline was predictive of fulfilment of RA according to the 2010 ACR/EULAR criteria after follow-up. We also confirmed that ERK activation is enhanced in patients with RA who develop erosive disease. Finally, with the new criteria we identified a subset of patients that fulfil 2010 ACR/EULAR criteria for RA at baseline, but have low levels of JNK activity and self-limiting disease.
Synovial JNK phosphorylation has previously been detected in patients with longstanding RA, and JNK contributes to interleukin 1β-induced collagenase expression, inflammation and joint destruction in murine arthritis models.12 18 41 Additionally, JNK signalling drives collagenase-3 expression and bone damage in rat adjuvant-induced arthritis.12 These studies have indicated a role for JNK in regulating synovial ECM biology, or cell interactions with the ECM. Comparing patients with early arthritis with self-limiting disease versus those with persistent disease, either non-erosive or erosive, we find that enhanced JNK activation is associated with decreased expression of developmentally important ECM components, including several forms of collagen, laminin C1 and MMP-12. We also find that CD44 expression is elevated in patients with early arthritis who develop persistent disease. CD44 is thought to have an important role in the pathology of RA, acting as an adhesion molecule and ECM component ligand for endothelial cells, macrophages, lymphocytes and FLS.42 Specific splice variants of CD44 are associated with invasive FLS behaviour in RA.43,–,46 Also, a pathogenic role for CD44 has been demonstrated in animal arthritis models.42 47 Further studies will determine if there is a direct relationship between expression of these genes and JNK activity in patients with early arthritis, and if so, whether JNK activation regulates or responds to these genes. Our studies indicate that evaluation of JNK activity might be of additional diagnostic and prognostic value to the new classification criteria, and that pharmacological targeting of JNK and ERK may be of benefit in limiting inflammation and joint destruction early in the development of RA.
Acknowledgments
The authors would like to thank Ms Beatrice M Fernandez, Ms LM Hartkamp and Ms IE van Es (University of Amsterdam) for assistance in mRNA preparation, qPCR array gene analysis and immunohistochemical studies.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
DdL and MGHvdS contributed equally to this manuscript.
PPT and KAR contributed equally to this manuscript.
-
Funding This research was supported by the Dutch Arthritis Association (NR 04-1-301). Funding was also provided by the European Community's FP6 funding (Autocure). This publication reflects only the views of the authors and the European Community is not liable for any use that may be made of the information herein.
-
Provenance and peer review Not commissioned; externally peer reviewed.