Article Text

Abstract
Objectives: The phase III RADIATE study examined the efficacy and safety of tocilizumab, an anti-IL-6 receptor monoclonal antibody in patients with rheumatoid arthritis (RA) refractory to tumour necrosis factor (TNF) antagonist therapy.
Methods: 499 patients with inadequate response to one or more TNF antagonists were randomly assigned to receive 8 mg/kg or 4 mg/kg tocilizumab or placebo (control) intravenously every 4 weeks with stable methotrexate for 24 weeks. ACR20 responses, secondary efficacy and safety endpoints were assessed.
Results: ACR20 was achieved at 24 weeks by 50.0%, 30.4% and 10.1% of patients in the 8 mg/kg, 4 mg/kg and control groups, respectively (less than p<0.001 both tocilizumab groups versus control). At week 4 more patients achieved ACR20 in 8 mg/kg tocilizumab versus controls (less than p = 0.001). Patients responded regardless of most recently failed anti-TNF or the number of failed treatments. DAS28 remission (DAS28 <2.6) rates at week 24 were clearly dose related, being achieved by 30.1%, 7.6% and 1.6% of 8 mg/kg, 4 mg/kg and control groups (less than p = 0.001 for 8 mg/kg and p = 0.053 for 4 mg/kg versus control). Most adverse events were mild or moderate with overall incidences of 84.0%, 87.1% and 80.6%, respectively. The most common adverse events with higher incidence in tocilizumab groups were infections, gastrointestinal symptoms, rash and headache. The incidence of serious adverse events was higher in controls (11.3%) than in the 8 mg/kg (6.3%) and 4 mg/kg (7.4%) groups.
Conclusion: Tocilizumab plus methotrexate is effective in achieving rapid and sustained improvements in signs and symptoms of RA in patients with inadequate response to TNF antagonists and has a manageable safety profile.
Trial registration number: NCT00106522.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Rheumatoid arthritis (RA) is a chronic, immune-mediated, systemic disease affecting approximately 1% of the population.1 It is characterised by pain, swelling and progressive destruction of the small joints of the hands and feet, accompanied by loss of function, fatigue, anaemia and an increased risk of osteoporosis and coronary heart disease.
Treatments often include disease-modifying antirheumatic drugs (DMARD; eg, methotrexate) and biologicals (eg, tumour necrosis factor (TNF) alpha inhibitors). However, even with TNF inhibitors (alone or with DMARD), 20–40% of RA patients show inadequate response. In addition, the attrition rate after 2 years nears 20%2 Switching between anti-TNF treatments, rising patient age and increasing disease duration all increase patients’ chances of an inadequate response.3 – 9 This partly explains the poor prognosis for, and the difficulty in, treating this population.
An alternative target for RA treatment is the pleiotropic cytokine IL-6. Chronic joint inflammation in RA leads to the production of IL-6 and its receptor, IL-6R, which is expressed on effector cells that cause and prolong inflammation. IL-6 also influences B and T-cell development, along with the activation of cells involved with the innate immune response.10 ,11 IL-6 knockout mice have been shown to be protected from developing joint symptoms in an arthritis model in vivo.12 ,13
Tocilizumab is a humanised anti-IL-6R monoclonal antibody that prevents IL-6 from binding to IL-6R.14 Tocilizumab in combination with methotrexate or DMARD exhibits superior clinical efficacy compared with controls in several populations, including patients with an inadequate response to methotrexate and/or DMARD.15 – 19 The Research on Actemra Determining effIcacy after Anti-TNF failurEs (RADIATE) study examined the efficacy and safety of tocilizumab with methotrexate in patients with active RA who had failed at least one TNF antagonist.
PATIENTS AND METHODS
Patients
Patients 18 years of age and older with moderate to severe active RA and failure to respond or intolerance to one or more TNF antagonists within the past year were included. Patients had active RA for 6 months or more, swollen joint count (SJC) of 6 or more, tender joint count (TJC) of 8 or more, and C-reactive protein (CRP) greater than 1.0 mg/dl or erythrocyte sedimentation rate (ESR) greater than 28 mm/h at baseline. Patients discontinued etanercept (⩾2 weeks), infliximab or adalimumab (⩾8 weeks), leflunomide (⩾12 weeks) and all DMARD other than methotrexate before receiving study medication. Patients had to be treated with methotrexate for 12 weeks or more before baseline (stable dose ⩾8 weeks).
Exclusion criteria included treatment with cell-depleting agents, uncontrolled medical conditions, history of other inflammatory diseases or functional class 4 RA, history of malignancies or recurrent infections, primary or secondary immunodeficiency, haemoglobin less than 8.5 g/dl, leucopenia, neutropenia, thrombocytopenia, abnormal liver function, triglycerides greater than 10 mmol/l, or recognised active tuberculosis, hepatitis B, or hepatitis C.
Study design
RADIATE was a phase III, randomised, double-blind, placebo-controlled, parallel group study conducted throughout North America and western Europe. Protocol approval by institutional review boards, ethics committees and/or regulatory authorities and written informed consent from each patient were obtained as per the Declaration of Helsinki. Participants were randomly assigned to tocilizumab 8 mg/kg or 4 mg/kg intravenously every 4 weeks or placebo intravenously every 4 weeks (controls). Drug/placebo was infused for one hour. All patients received stable methotrexate (10–25 mg weekly) and folate. No other DMARD were allowed. Patients were allowed to continue stable oral corticosteroids (⩽10 mg/day prednisone or equivalent) and/or non-steroidal anti-inflammatory drugs.
Rescue therapy of 8 mg/kg tocilizumab plus methotrexate was offered at week 16 in all cases of treatment failure (<20% improvement in both SJC and TJC). Joint assessors were blinded as to other data including CRP, ESR and treatment assignment, thus rescue therapy could be given to patients already receiving 8 mg/kg tocilizumab.
Study endpoints
The primary endpoint was the ACR20 response at week 24. Secondary endpoints included further efficacy measures. Safety outcomes included adverse events, infections and infusion reactions.
Statistical methodology
A sample size of 450 patients was calculated to provide more than 80% power to detect a difference of 20 points between tocilizumab and control arms at week 24 for the ACR20 response and to enable the reporting of safety and efficacy for this unique patient population for registration. Primary endpoint analysis was performed on all participants receiving one or more administration of study treatment (the intent to treat (ITT) population). Safety data are presented using the safety population, comprising all ITT patients with one or more post-randomisation assessments of safety.
Primary endpoint analysis for ACR20 response (with secondary analyses for ACR50/70, DAS28 and European League Against Rheumatism (EULAR) responses) compared the proportion of patients in each of the tocilizumab plus methotrexate groups versus controls with a response at week 24 using a Cochrane–Mantel–Haenszel χ2 test with adjustment for site. Patients on rescue therapy or with insufficient data to calculate the change from baseline ACR score at a specific time point were classified as non-responders at that time point. Changes from baseline in the individual ACR core set parameters and haemoglobin were summarised using descriptive statistics, and the difference between treatment groups for each component was compared using an analysis of variance with adjustment for site.
In the case of missing data, SJC and TJC scores were calculated as the last observation carried forward. There was no imputation for missing data for the remaining ACR components. Comparisons between tocilizumab groups and for safety outcomes were not performed.
RESULTS
Patients
The three groups were reasonably well balanced for demographics and RA characteristics at baseline (table 1).
Patient disposition is shown in fig 1. The major reason for screen failure was a CRP or ESR value below the inclusion limit. This did not skew the populations, which were well balanced. One control patient withdrew after random selection but before receiving any study treatment because of a latex allergy. Also, one patient randomly assigned to the 4 mg/kg plus methotrexate group received 6 mg/kg of tocilizumab at baseline. This patient was assigned to the 8 mg/kg group for safety analyses and to the 4 mg/kg group for ITT analyses but was excluded from per protocol analyses. The most common reasons for withdrawal were: adverse events in 11 (8 mg/kg), 10 (4 mg/kg) and 10 (control) and insufficient therapeutic response in four, six and 19 patients, respectively. More patients in the control group (41%) and in the 4 mg/kg group (19%) received rescue therapy after week 16 compared with 11% of patients in the 8 mg/kg group.

Efficacy
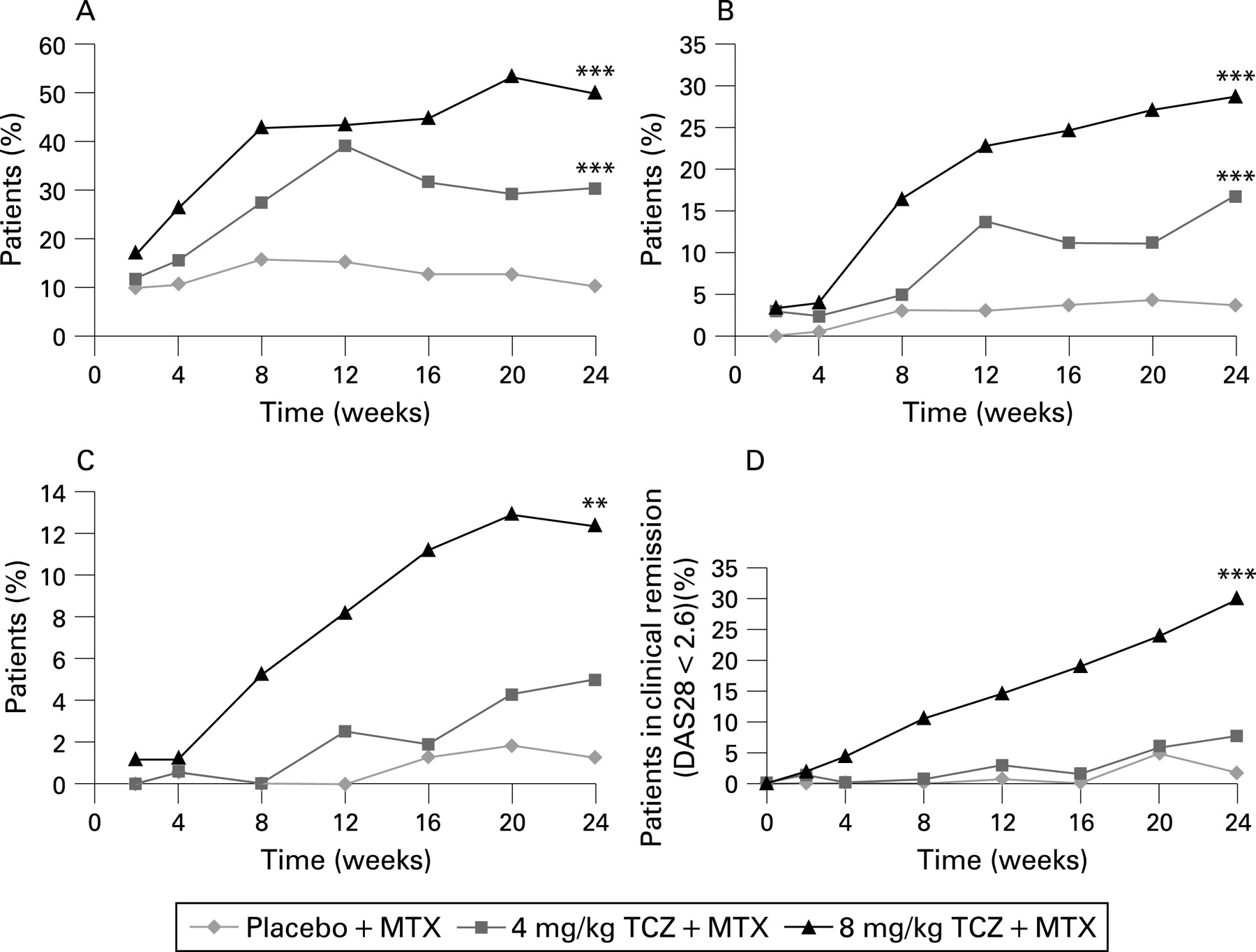
Both the 8 mg/kg (50.0%) and 4 mg/kg (30.4%) groups exhibited superior ACR20 responses compared with control (10.1%; less than p<0.001). ACR50 and ACR70 responses at 24 weeks were achieved by 28.8% and 12.4% of patients in the 8 mg/kg group (less than p<0.001 and less than p = 0.001, respectively, versus control), 16.8% and 5.0% in the 4 mg/kg group (less than p<0.001 and p = 0.1, respectively, versus control) and 3.8% and 1.3% in the control group. Response to tocilizumab treatment was rapid (fig 2A–C). The ACR20 response was comparable irrespective of the type or number of failed TNF antagonists (table 2). Ninety-five per cent of previous TNF antagonist failures were due to inadequate efficacy.

Significant improvements from baseline were observed at 24 weeks for SJC: −7.8 versus −6.8 versus −0.5 (less than p<0.001 versus control for both tocilizumab groups) and TJC: −14.8 versus −10.5 versus 0.3 in the 8 mg/kg, 4 mg/kg and control groups, respectively (less than p<0.001 versus control for both tocilizumab groups). Likewise, Health Assessment Questionnaire (HAQ) values improved from baseline by −0.39 versus −0.31 versus −0.05 in the 8 mg/kg, 4 mg/kg and control groups, respectively (less than p<0.001 for control versus 8 mg/kg and p = 0.003 versus 4 mg/kg group).
Low disease activity (DAS28 <3.2) was reported at week 24 in 51.2%, 15.2% and 4.9% of patients in the 8 mg/kg, 4 mg/kg and control groups, respectively. DAS28 remission (DAS28 <2.6) rates at 24 weeks were 30.1%, 7.6% and 1.6% in the 8 mg/kg, 4 mg/kg and control groups (less than p = 0.001 (8 mg/kg), p = 0.053 (4 mg/kg) both versus control). The dose response between 8 mg/kg and 4 mg/kg is evident, as seen by the proportion of patients achieving DAS28 remission (DAS28 <2.6) as early as week 4 (fig 2D). Furthermore, there was an incremental improvement in this measure in the 8 mg/kg group at each time point throughout the 24 weeks.
Good or moderate EULAR responses at week 24 were observed in 67.7%, 46.5% and 16.5% of the 8 mg/kg, 4 mg/kg and control groups, respectively (less than p<0.001 across all categories for both tocilizumab groups versus control).
CRP levels and ESR dropped markedly by week 2 in both tocilizumab groups (fig 3A–B). By week 24, mean CRP had normalised (<0.3 mg/dl) in the 8 mg/kg group, but not in the 4 mg/kg or control groups. Mean haemoglobin levels increased with tocilizumab as early as week 2, more with 8 mg/kg, with incremental improvements over time (fig 3C).

{kind=link}
{kind=link}
{kind=link}
Safety
The overall incidence of adverse events was similar in all groups (table 3). Adverse events were mostly mild to moderate, with more attributed to study treatment in the tocilizumab groups. There was no obvious influence of the previous type or number of TNF antagonist treatments (data not shown).
There were more serious adverse events in the control group compared with the individual tocilizumab treatment arms (table 3). These were primarily related to complications of RA. The incidence of serious related adverse events was also higher in the 8 mg/kg group (five) than controls (three). The rate of serious infections per 100 patient-years was 9.98, 5.72 and 9.64 in the 8 mg/kg, 4 mg/kg and control groups, respectively. All but two serious infections, staphylococcal polyarthritis in the 8 mg/kg group and cellulitis in the control group, resolved without sequelae. Five serious infections in four patients led to study treatment discontinuation: staphylococcal polyarthritis in the 8 mg/kg group; necrotising pneumonia in the 4 mg/kg group; urosepsis and osteomyelitis/cellulitis in the controls.
Infusion reactions (adverse events occurring during or within 24 h after infusion) occurred in 9.1% (8 mg/kg), 9.8% (4 mg/kg) and 6.3% (control) of patients, resulting in two withdrawals: one control patient and one in the 8 mg/kg group. Most reactions were transient non-specific symptoms such as headache or hypertension; none were consistent with anaphylaxis.
Mean neutrophil counts decreased within the normal range in patients receiving tocilizumab plus methotrexate. Using common toxicity criteria grades (CTC, version 2.0) to classify neutropenia, 49 patients (28.0%), 33 patients (20.3%) and one patient (<1.0%) in the 8 mg/kg, 4 mg/kg and control groups, respectively, had transient neutropenia at some time during the study. Although most changes were CTC grade 1 or 2, four patients in the 8 mg/kg group and one in the 4 mg/kg group had grade 4 neutropenia (absolute neutrophil count <500) requiring withdrawal per protocol. One of the patients in the 8 mg/kg group was inadvertently entered into the study with grade 4 neutropenia and was withdrawn immediately after the first dose. In addition, five patients (four in the 8 mg/kg group, one in the 4 mg/kg group) had transient grade 3 neutropenia. No patients had associated fever or serious infection.
Although transient or recurrent increases in alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) above the upper limit of normal (ULN; 55 U/l) were common as patients were also receiving methotrexate, no patients developed evidence of methotrexate hepatotoxicity. Increases in ALT from normal at baseline to more than three times ULN to five times ULN occurred in four patients (2%) in the 8 mg/kg group, four patients (2.5%) in the 4 mg/kg group and one control patient (<1%). None of these elevations were sustained for more than two consecutive visits. In most patients, values normalised while continuing tocilizumab therapy. Per protocol, three patients discontinued study treatment for elevated liver enzymes. Mild to moderate hepatic steatosis diagnosed by imaging methods was recorded in one patient each in the 8 mg/kg and 4 mg/kg groups, without clinically apparent hepatitis or hepatic dysfunction.
Elevations in high-density lipoprotein and low-density lipoprotein, as defined by ATPIII guidelines,20 are shown in table 3. Clinically relevant triglyceride increases without evidence of pancreatitis were observed in two patients in the 4 mg/kg group. Comparable proportions of patients had a greater than 30% increase in the ApoB/ApoA atherogenic index: 11.6% (8 mg/kg), 9.4% (4 mg/kg) and 9.7% (controls). More patients in the tocilizumab groups had a greater than 30% increase in the low-density/high-density lipoprotein index: 22.2% (8 mg/kg), 19.1% (4 mg/kg) and 10.1% (controls). During the study, there were no reported ischaemic cardiac disorders in tocilizumab-treated patients. There was only one cardiovascular event (myocardial infarction) in the control group.
DISCUSSION
RADIATE is the first study demonstrating the efficacy of tocilizumab plus methotrexate in patients with an inadequate response to TNF antagonist treatment. Treatment with tocilizumab, especially at the 8 mg/kg dose, and stable methotrexate provides a rapid and sustained improvement in RA symptoms. Treatment was generally well tolerated, with a comparable incidence of serious infections and withdrawals due to safety issues. These results were consistent with the results obtained in previous trials in different patient populations, including the recently reported OPTION and TOWARD trials.15 ,19 ,21 As observed in those trials, the onset of ACR, DAS28 and EULAR responses in RADIATE occurred within 2–4 weeks of tocilizumab 8 mg/kg treatment.
Patients receiving 8 mg/kg tocilizumab plus methotrexate exhibited the greatest ACR20/50/70 responses at 24 weeks; all numerically higher compared with 4 mg/kg and significantly higher than control, irrespective of the most recently failed, or number of failed anti-TNF therapies.
Treatment goals for RA have shifted towards achieving remission. Similar to OPTION and TOWARD, in this study over half of patients receiving 8 mg/kg tocilizumab achieved low disease activity (DAS28 <3.2) and nearly a third achieved DAS28 remission (DAS28 <2.6) after 24 weeks of treatment. This is somewhat higher than the ACR50 response rate and may relate to the substantial suppression of acute phase reactants by tocilizumab treatment. DAS28 scores continued to improve linearly throughout the 24 week period in the 8 mg/kg tocilizumab group, which suggests that DAS28 remission rates may continue to improve with longer treatment periods. DAS28 improvement was associated with incremental decreases in not only ESR and CRP, beginning by week 2, but also with improvements in patient global health, SJC and TJC, from week 4 onwards (data not shown). Collectively, the efficacy data support the use of 8 mg/kg tocilizumab plus methotrexate in this patient population, and together with improvements in HAQ, provide evidence that changing therapy from an anti-TNF biological to IL-6R inhibition with tocilizumab can improve RA signs and symptoms in inadequate responders to anti-TNF therapy.
The accumulation of IL-6 and soluble IL-6R prolongs the inflammatory response and produces a variety of systemic conditions that are commonly seen in RA, including anaemia of chronic inflammation.22 ,23 Treatment that reduces the severity of inflammation has been associated with an increase in haemoglobin levels.22 Indeed, the concomitant reduction in CRP and increase in haemoglobin observed in this study in patients receiving 8 mg/kg tocilizumab supports this hypothesis. A proposed mechanism for the increase in haemoglobin levels relates to the ability of IL-6 to induce hepcidin production as part of the acute phase response.24 IL-6 signalling activates the JAK–STAT pathway to stimulate the production of acute phase proteins, including CRP and hepcidin. Because hepcidin blocks iron transport, inhibition of hepcidin production should lead to the release of sequestered iron in macrophages and the alleviation of anaemia. TNF does not stimulate hepcidin production.25 ,26 The correction of anaemia seen with TNF antagonists thus potentially occurs indirectly via a reduction in IL-6 levels,27 whereas tocilizumab directly inhibits IL-6 signalling for each mechanism.
Although numerically higher in the control group, the incidence of adverse events and serious adverse events was comparable across treatment groups and most adverse events were mild or moderate in intensity. Infections were the most frequently reported adverse event. The rate of serious infections was similar in the 8 mg/kg and control groups and was consistent with the OPTION study.19 These rates are higher than reports in other similar studies; however, they are comparable to rates in a recent large community study and reflect the comorbidities and long disease duration seen in this treatment population,28 – 30 as well as concomitant corticosteroid treatment in more than half of the patients. Four patients discontinued study treatment due to serious infections, one or two from each study group. No tuberculosis or opportunistic infections were observed.
Whereas a higher proportion of patients experienced gastrointestinal adverse events in the 8 mg/kg group, only two patients in each treatment group had serious gastrointestinal adverse events. One patient in the 8 mg/kg group with a history of diverticulosis had an acute diverticular perforation 5 days after her second infusion and recovered fully. Tocilizumab may suppress symptoms and delay the detection of diverticulitis; thus patients with a history of diverticulitis must be managed carefully.
Patients treated with tocilizumab in the present study demonstrated reductions in mean neutrophil counts. Although neutropenia was typically transient, not associated with infection or fever and of low CTC grade, more patients in this population (TNF antagonist inadequate responders) had transient grade 3 or 4 neutropenia, compared with reports in the methotrexate inadequate responder population of the OPTION study.19 Some possible mechanisms by which tocilizumab may result in lower neutrophil counts include blocking IL-6-induced neutrophil survival, the downregulation of other inflammatory cytokines and the margination of neutrophils from the circulation into tissues.31 – 34
Consistent with previous reports, tocilizumab treatment in this study was associated with episodic increases in hepatic aminotransferases. No patient had both a greater than three times ULN increase in ALT and AST and a clinically relevant (greater than two times ULN) increase in total bilirubin and there were no cases of hepatitis or clinically significant hepatic dysfunction. Nonetheless, long-term follow-up is required to determine the implications of these observations.
Increases in total and low-density lipoprotein cholesterol levels in this study were consistent with previous reports17 ,19 ,35 and were associated with marked decreases in CRP. Patients with active RA often have abnormal lipid profiles, an inverse relationship that associates low lipid levels with elevated acute phase reactants in some patients.36 ,37 Considering the recognised higher incidence of cardiovascular mortality in RA patients,37 ,38 – 41 traditional risk factors for cardiovascular disease, including dyslipidaemia, are not necessarily predictive of events for patients with RA. Indeed, several studies have provided evidence that despite increases in lipid levels, reduced inflammation markers have been associated with reduced cardiovascular mortality.38 ,40 – 43 Interestingly, the ApoB/ApoA ratio is viewed as possibly the more relevant indicator for inflammatory vascular disorders than other atherogenic indices, and this index worsened in a comparable proportion (∼10%) of patients in each treatment group in this study.44 ,45
The current study does have limitations. First, the clinical consequences of the increase in lipid levels observed with 24-week tocilizumab treatment, together with the significant decreases in CRP and inflammation, are unclear. Long-term studies in this population may determine whether the effects of tocilizumab on lipid levels are clinically meaningful, although it is apparent that this effect may require treatment with statins, according to standards developed to lower cardiovascular risk. Second, the limitation of joint damage with tocilizumab treatment (as shown in the SAMURAI study)18 needs to be confirmed in patients refractory to anti-TNF treatments.
CONCLUSION
In patients with moderate to severe active RA responding inadequately or who are intolerant to TNF antagonists, changing to tocilizumab plus methotrexate is effective, and the safety profile is manageable, regardless of the number of previous failed agents.
Acknowledgments
The authors acknowledge the collaboration and commitment of all investigators and their staff. Dr Thasia Woodworth provided added analysis and interpretation of the data. Dr Gordon Brooker, Phocus Services, Basel, Switzerland, provided editorial assistance.
REFERENCES
Footnotes
-
Funding: Grant support was received from F Hoffmann-La Roche Ltd and Chugai Pharma KK.
-
Competing interests: PE, EK, RvV and JK have received research grants from F Hoffmann-La Roche Ltd. PE, RvV and JK have received consulting fees from F Hoffmann-La Roche Ltd. EA and JL are employees of Roche Products.
-
Ethics approval: Protocol approval by institutional review boards, ethics committees and/or regulatory authorities was obtained.
-
Patient consent: Obtained.