Article Text

Abstract
Objectives The aim of this study was to identify the role of tenascin-C (TNC) in entheseal new bone formation and to explore the underlying molecular mechanism.
Methods Ligament tissue samples were obtained from patients with ankylosing spondylitis (AS) during surgery. Collagen antibody-induced arthritis and DBA/1 models were established to observe entheseal new bone formation. TNC expression was determined by immunohistochemistry staining. Systemic inhibition or genetic ablation of TNC was performed in animal models. Mechanical properties of extracellular matrix (ECM) were measured by atomic force microscopy. Downstream pathway of TNC was analysed by RNA sequencing and confirmed with pharmacological modulation both in vitro and in vivo. Cellular source of TNC was analysed by single-cell RNA sequencing (scRNA-seq) and confirmed by immunofluorescence staining.
Results TNC was aberrantly upregulated in ligament and entheseal tissues from patients with AS and animal models. TNC inhibition significantly suppressed entheseal new bone formation. Functional assays revealed that TNC promoted new bone formation by enhancing chondrogenic differentiation during endochondral ossification. Mechanistically, TNC suppressed the adhesion force of ECM, resulting in the activation of downstream Hippo/yes-associated protein signalling, which in turn increased the expression of chondrogenic genes. scRNA-seq and immunofluorescence staining further revealed that TNC was majorly secreted by fibroblast-specific protein-1 (FSP1)+fibroblasts in the entheseal inflammatory microenvironment.
Conclusion Inflammation-induced aberrant expression of TNC by FSP1+fibroblasts promotes entheseal new bone formation by suppressing ECM adhesion forces and activating Hippo signalling.
- arthritis
- experimental
- chondrocytes
- fibroblasts
- inflammation
- spondylitis
- ankylosing
Data availability statement
Data are available upon reasonable request. The data that support the findings in this study are available from the corresponding author upon request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
No targeted and effective treatments have been developed to satisfactorily prevent pathological new bone formation in ankylosing spondylitis (AS).
Tenascin-C (TNC) is an extracellular matrix protein upregulated in multiple inflammatory conditions. TNC protein synthesis is tightly regulated with restricted distribution in adult tissues.
What does this study add?
TNC is aberrantly upregulated in enthesis and ligament tissues in patients with AS and animal models.
Genetic ablation and pharmacological inhibition of TNC suppress entheseal new bone formation in animal models.
Inflammation-induced aberrant expression of TNC by fibroblast-specific protein-1+fibroblasts promotes entheseal new bone formation through suppression of extracellular matrix adhesion force and activation of Hippo signalling.
How might this impact on clinical practice or future developments?
Suppression of aberrant expression of TNC may be a potential therapeutic strategy for prevention of pathological new bone formation in AS.
Introduction
Axial spondyloarthritis (SpA) is a chronic inflammatory disease that mainly affects the axial skeleton and has a global prevalence of 0.32%–1.4%.1 2 SpA includes non-radiographic SpA and radiographic axial SpA, which is also termed ankylosing spondylitis (AS).1 In addition to inflammatory back pain, spinal ankylosis and immobilisation resulting from entheseal pathological new bone formation are typical features of AS.3 Given that the affected population is mainly young and middle-aged men, disability caused by AS is a burden to the patients and society, resulting in considerable socioeconomic costs.4
Although recent investigations and medications have focused on the suppression of inflammation and pain control, treatment targeting pathological bone formation is lacking, and the prognosis of axial structural damage remains unsatisfactory.5 The pathogenesis of entheseal pathological new bone formation that consequently leads to bony bridging is not well understood. Although some molecules that are critical for bone formation have been hypothesised in the mechanism of AS, including bone morphogenetic proteins,6 Dickkopf-17 8 and Wnt proteins,9 directly targeting these molecules and related pathways of general bone metabolism might be associated with excessive side effects. Therefore, more precise therapeutic targets that merely function in pathological process with less negative effect on physiological function are needed for the treatment of spinal ankylosis.
Tenascin-C (TNC) is a large molecular extracellular matrix (ECM) glycoprotein hexameric multidomain protein. Upregulation of TNC is noted in multiple inflammatory conditions, including traumatic injuries or light-damaged skin, bacterial infections and asbestos-induced damaged lungs.10–17 Recent investigations have also reported increase of serum level of TNC in patients with rheumatic diseases,18 including systemic lupus erythematosus, psoriatic arthritis and AS.19 However, whether TNC plays a role in the process of entheseal pathological new bone formation is unknown.
In the current study, we found that TNC was aberrantly upregulated in ligament and entheseal tissues from patients with AS and animal models. Systemic neutralisation with specific antibody or genetic ablation of TNC significantly suppressed entheseal pathological new bone formation in animal models. Therefore, TNC might be essential for the development of pathological new bone formation. Investigation of the role of TNC in the process of pathological new bone formation and the underlying molecular mechanism might shed more light on the enigma of axial skeleton ankylosis and propose a potential therapeutic direction for the disease.
Materials and methods
Additional detailed information is provided in online supplemental file.
Supplemental material
Patients
With ethics committee approval and patient consent, the samples (bone, ligamentum flavum, supraspinatus ligament and interspinous ligament) involved in spinal ankylosis from patients with AS and non-AS patients were collected during surgeries. Twenty-two patients (10 with AS and 12 with non-AS) were enrolled between September 2015 and June 2019. The indications of surgery for patients with AS included disabling kyphosis, loss of horizontal vision without compensation, painful spinal pseudarthrosis or Andersson lesion.20 Non-AS patients without any systemic inflammatory condition including SpA fulfilled the indications for correction of scoliosis or spinal decompression of thoracic or lumbar stenosis.21–23
Mice
DBA/1 and C57BL/6J mice were purchased from the Charles River Laboratories. The TNC knockout (KO) mouse model (C57BL/6J) was created by Cyagen Biosciences via using CRISPR/Cas-mediated genome engineering. Exon 3–5 of TNC gene (NCBI Reference Sequence: NM_011607.3; Ensembl: ENSMUSG00000028364) were selected as target site.
For spontaneous arthritis model, male DBA/1 mice (8 weeks) were mixed and caged together in groups of nine mice to induce arthritis. For antibody administration, the mice received treatment intraperitoneally once a week with TNC-neutralising antibody (5 mg/kg) (MAB2138, R&D Systems) or the equivalent volume of vehicle antibody (MAB006, R&D Systems) since the second week after caging. For Hippo pathway signalling inhibition, the mice received treatment intraperitoneally three times a week with XMU-MP-1 (2 mg/kg) (HY-100526, a reversible and selective MST1/2 inhibitor, MedChemExpress) since the second week after caging. Dimethyl sulfoxide (DMSO) was administered as a negative control.
For collagen antibody-induced arthritis (CAIA) model, wild-type (WT) and TNC KO C57BL/6J mice (male, 10 weeks) were injected intraperitoneally with Arthrogen-CIA monoclonal antibody cocktail (4 mg/20 g) (Chondrex) on day 0. Then, 100 µg lipopolysaccharide (LPS) was injected intraperitoneally into each mouse on day 3. The equivalent volume of non-specific immunoglobulin (day 0) and LPS (day 3) were used for control purposes.
At the end of each experimental time point, mice were sacrificed. Hind paw specimens were dissected and fixed with 4% paraformaldehyde for μCT and histological analyses.
Statistical analysis
Statistical analyses were performed using SPSS V.22.0. All data obtained from experiments repeated at least three times was represented as mean±SD. Differences between two groups were analysed using two-tailed Student’s t-test. Comparisons of multiple groups were analysed via one-way analysis of variance. The level of significance was set at p<0.05.
Results
TNC is upregulated in ligament tissues from patients with AS and animal models
To investigate the molecular mechanism of pathological new bone formation in AS, the spinal ligament tissues were collected from patients with AS and age-matched and sex-matched controls who underwent correction surgeries (figure 1A, online supplemental figure S1A). Immunohistochemical staining showed infiltration of CD68+macrophages and expression of TNFα and IL-17A in the tissue samples from patients with AS, indicating an inflamed status (online supplemental figure S1B‒D). RNA sequencing analyses showed significant enrichment of ECM-related GO terms including Extracellular Matrix Organisation (GO:0030198) and Extracellular Structure Organisation (GO:0043062) in differentially expressed genes (figure 1B). The differentially upregulated genes of these two GO terms were selected for further study (figure 1C). Gene Set Enrichment Analysis showed a high normalised enrichment score of the TNC_TARGETS gene set (figure 1D,E). Immunohistochemical staining and western blot analysis confirmed that TNC was upregulated at the entheses of spinal ligament tissues from patients with AS (figure 1F,G, online supplemental figure S1E). Two standard animal models that mimic the pathological features of entheseal pathological new bone formation of AS were established.24–29 Entheseal pathological new bone formation was confirmed by μCT (online supplemental figure S2A‒F) and histological staining (online supplemental figure S2G,H). In accordance with the findings from human tissues, TNC was also aberrantly upregulated at the entheseal site of the hind paws in these two animal models (figure 1H‒K).
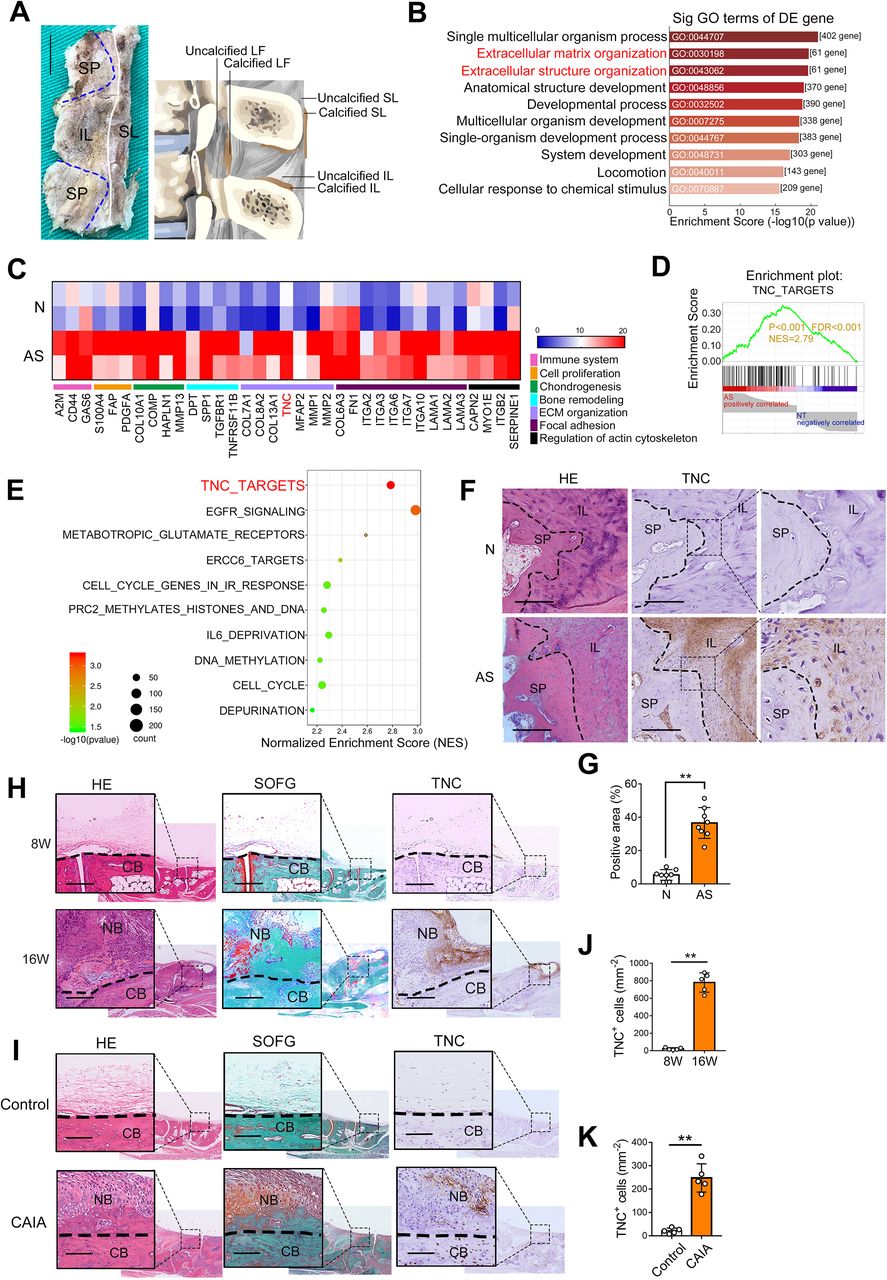
TNC is upregulated in ligament tissues from patients with AS and animal models. (A) An illustration of spinal ligament tissues collection. Scale bar: 1 cm. (B) GO terms-enriched analysis of differentially expressed genes in AS entheseal tissues (top 10 significant). (C) Heat map of differentially upregulated genes from Extracellular Matrix Organization (GO:0030198) and Extracellular Structure Organization (GO:0043062). (D) Representative GSEA results for TNC_TARGETS gene set. (E) Significantly enriched signalling pathways of GSEA pathway enrichment analysis. (F,G) H&E staining, immunohistochemical analysis and quantitative analysis of TNC and in entheseal tissues from patients with AS and non-AS patients. Scale bar: 300 µm. n=8 per group. (H) H&E staining, Safranin O Fast Green (SOFG) staining and immunohistochemical analysis of TNC in hind paws of male DBA/1 model. Scale bar: 200 µm. (I) H&E staining, SOFG staining and immunohistochemical analysis of TNC in hind paws of CAIA model. Scale bar: 100 µm. (J) Quantitative analysis of TNC-positive cells (mm−2) in (H). n=5 per group. (K) Quantitative analysis of TNC-positive cells (mm−2) in (I). n=5 per group. Data are presented as mean±SD. **p<0.01, unpaired t-test. AS, ankylosing spondylitis; CAIA, collagen antibody-induced arthritis; CB, cortical bone; ECM, extracellular matrix; FDR, false discovery rate; GSEA, Gene Set Enrichment Analysis; IL, interspinous ligament; LF, ligamentum flavum; N, non-AS patients; NB, new bone; SL, supraspinous ligament; SP, spinous process; TNC, tenascin-C.
Inhibition of TNC suppresses entheseal pathological new bone formation
To confirm the critical role of TNC in pathological new bone formation, a TNC-neutralising antibody was administered systemically to DBA/1 mice model 2 weeks after caging and to CAIA model 7 days after immunisation. The results showed that pathological bone formation was significantly suppressed in TNC antibody-treated group, as determined by μCT analysis, H&E staining and Safranin O Fast Green (SOFG) staining (figure 2A‒D, online supplemental figure S3A,B). Furthermore, a CAIA model was established in TNC KO mice. The successful creation of TNC KO mice was validated before induction (online supplemental figure S4). TNC KO mice were born alive. Consistent with previous reports, they show no abnormalities physiologically.30 No significant difference was observed in the appearance and weight of these KO mice compared with the WT mice. However, they did show some abnormal behaviours as reported, such as moving about their cages almost incessantly regardless of the dark-light cycle.31 The results showed that the development of pathological new bone was dramatically suppressed in TNC−/− CAIA mice, as detected by μCT analysis, H&E staining and SOFG staining (figure 2E,F).

Inhibition of TNC suppresses entheseal pathological new bone formation. (A) μCT images and quantitative analysis of pathological new bone formation in male DBA/1 model. Red arrows indicate the new bones. n=9 per group. Scale bar: 1 mm. (B) H&E staining and Safranin O Fast Green (SOFG) staining in hind paws of male DBA/1 model at the age of 24 w. Scale bar: 200 µm. (C) μCT images and quantitative analysis of pathological new bone formation in CAIA model. Red arrows indicate the new bone. n=5 per group. Scale bar: 1 mm. (D) H&E staining and SOFG staining in hind paws of CAIA model. Scale bar: 200 µm. (E) μCT images and quantitative analysis of hind paws of control mice and CAIA mice with and without TNC knockout. Red arrows indicate the new bone. n=5 per group. Scale bar: 1 mm. (F) H&E staining and SOFG staining in hind paws of control mice and CAIA mice with and without TNC knockout. Scale bar: 100 µm. Data are presented as mean±SD. *p<0.05, **p<0.01, determined by unpaired, two-tailed Student’s t-test. Ab, antibody; BV, bone volume; CAIA, collagen antibody-induced arthritis; CB, cortical bone; NB, new bone; TNC, tenascin-C; Veh, vehicle.
TNC is critical for chondrogenesis in the process of endochondral ossification
Cartilage formation was observed in entheseal tissues from both patients with AS and animal models (online supplemental figure S5A,B), which was in accordance with previous reports indicating endochondral ossification as the major mechanism of entheseal pathological new bone formation.32 Our results further showed that genetic ablation and pharmacological inhibition of TNC had a suppressive effect on the formation of cartilage templates in both DBA/1 model and CAIA model (figure 3A,B, online supplemental figure S5C,D), indicating that TNC was involved in the process of endochondral ossification. Immunofluorescence staining revealed that TNC was expressed around chondrocytes (online supplemental figure S5E). In a cell culture system of human mesenchymal stem cells (hBMSCs) plated on TNC-coated dishes, chondrogenic differentiation was significantly enhanced, as detected by Alcian blue staining, qPCR and western blot (online supplemental figure S5F‒H). Moreover, the chondrogenic effect of TNC was suppressed by TNC antibody (figure 3C‒H). To confirm the chondrogenic effect of TNC in vivo, BMSCs isolated from TNC+/+ and TNC−/− mice were explanted into nude mice recipients (figure 3I). Ablation of TNC in BMSCs led to a significant reduction in chondrogenic potential (figure 3J). Collectively, these results suggest that TNC promotes chondrogenesis in the process of endochondral ossification.

TNC is critical for chondrogenesis in the process of endochondral ossification. (A) Immunofluorescence staining of Sox9 and Col10a1 in hind paws of control mice and CAIA mice with and without TNC knockout. Scale bar: 100 µm. (B) Quantitative analysis of Sox9-positive and Col10a1-positive cells (mm−2) in (A). n=3 per group. (C) Alcian blue staining of human mesenchymal stem cells planted on TNC-coated culture dish in micromass cultures with antibody administration for 7 days. (D,E) Western blot analysis and qRT-PCR analysis of the level of Sox9, Aggrecan, Collagen II in mesenchymal stem cells planted on TNC with antibody in micromass cultures for 48 hours. n=3. (F,G) Immunofluorescence staining of Sox9 (green) and Aggrecan (red) in human mesenchymal stem cells planted on TNC-coated culture dish in micromass cultures with antibody administration for 48 hours. Scale bar: 100 µm in (F). 50 µm in (G). (H) Mean fluorescence intensity of Sox9, Aggrecan in arbitrary units of (F,G). n=3. (I) Schematic diagram illustrating the experimental setup. (J) Safranin O staining and chondrogenic markers expression levels of cartilage-like tissues isolated from mice transplanted with TNC+/+ or TNC-/- MSCs. Scale bar: 50 µm. Data are presented as mean±SD. **p<0.01, *p<0.05, unpaired t-test. Ab, antibody; AS, ankylosing spondylitis; CAIA, collagen antibody-induced arthritis; CB, cortical bone; MSCs, mesenchymal stem cells; NB, new bone; TNC, tenascin-C; Veh, vehicle.
TNC promotes chondrogenesis by decreasing the matrix adhesion force
The modulation of ECM mechanical properties has been proven to regulate cell fate determination.33–37 To determine whether the chondrogenic effect of TNC was related to this underlying mechanism, the mechanical properties of tissues from both humans and CAIA model were investigated by atomic force microscopy (figure 4A). Results showed that the adhesion force was significantly decreased in tissues from patients with AS (figure 4B,C). Similarly, the adhesion force was also significantly decreased in entheseal tissues collected from CAIA model. However, this change was much less significant in TNC −/− CAIA model (figure 4D,E), suggesting that TNC deposition and its chondrogenic effect occurred along with a decrease in tissue adhesion force. To confirm that TNC was involved in decreasing the matrix adhesion force, different densities of RGD peptide were plated on Matrigel matrix with or without TNC (figure 4F). TNC significantly reduced the adhesion force of the matrix with RGD (figure 4G). The chondrogenesis of mesenchymal stem cells cultured on high adhesion matrix was suppressed compared with those cultured on low adhesion matrix (figure 4H,I); in the presence of TNC, this suppressive effect was alleviated (figure 4J). These results suggest that TNC promotes chondrogenesis by modulating ECM biomechanical properties, specifically, the adhesion force.

TNC promotes chondrogenesis by decreasing the matrix adhesion forces. (A) Schematic diagram illustrating the experimental setup. (B) AFM maps of ECM adhesion force in human samples from patients with AS and non-AS patients. (C) Graphs illustrate the adhesion force. n=5 per group. (D) AFM maps of ECM adhesion force in CAIA mice and control mice. (E) Graphs illustrate the adhesion force. n=5 per group. (F) Schematic diagram illustrating the experimental setup. (G) Adhesion force of coated Matrigel as described in (F). (H) Immunofluorescence image of Collagen II (green) in ADTC5 cells plated on Matrigel coated with low or high density RGD for 72 hours. (I) Western blot analysis of Sox9, Aggrecan, Col II protein levels in ADTC5 cells plated on Matrigel coated with low or high density RGD for 72 hours. (J) Alcian blue staining and western blot analysis of Sox9, Aggrecan, Collagen II protein levels in human mesenchymal stem cells in micromass cultures. Data are presented as mean±SD. **p<0.01, *p<0.05, unpaired t-test. AFM, atomic force microscopy; AS, ankylosing spondylitis; BSA, bovine serum albumin; CAIA, collagen antibody-induced arthritis; ECM, extracellular matrix; FV, force volume; RGD, Arg-Gly-Asp; TNC, tenascin-C.
TNC-mediated reduction in matrix adhesion force activates Hippo/YAP signalling
To investigate the downstream signalling of the TNC-mediated changes in the ECM adhesion force, RNA sequencing of the spinal ligament tissues from patients was conducted and pathway enrichment was analysed. The results revealed a significant enrichment of genes from Hippo signalling pathway (figure 5A). Immunohistochemistry staining showed that phosphorylation of yes-associated protein (YAP), which is an indicator of Hippo signalling activation, was upregulated at the sites where TNC was highly accumulated in the spinal ligament tissues from patients with AS (figure 5B). To confirm the activating effect of TNC on Hippo/YAP signalling, cells were plated on TNC-coated dishes under chondrogenic induction. As expected, the protein levels of pYAP and pLATS1 were upregulated with TNC-coated treatment (figure 5C), and nuclear translocation of YAP was therefore decreased (figure 5D). To further investigate whether TNC activates Hippo signalling through depolymerisation of actin cytoskeleton, lysophosphatidic acid (LPA), a bioactive lipid that promotes actin stress fibre formation,38 was applied to cells cultured under TNC-coated conditions (figure 5E). Western blot analysis showed that TNC-upregulated pYAP and pLATS1 were decreased by LPA (figure 5F), and nuclear translocation of YAP was therefore increased (online supplemental figure S6A‒C). These findings indicate that TNC is involved in actin cytoskeleton depolymerisation, subsequent Hippo pathway activation and YAP degradation.

TNC-mediated reduction in matrix adhesion force activates Hippo/YAP signalling. (A) Pathway enrichment analysis by Metascape of differentially expressed genes in patients with AS. (B) Immunohistochemical staining of phosphorylated YAP (S127) in entheseal tissues. Scale bar: 500 µm. (C,D) Protein levels of phosphorylated YAP (S127), total YAP, phosphorylated LATS1, total LATS1 and immunofluorescence image of YAP in ADTC5 cells plated on TNC for 6 hours. Ctrl: fibronectin 1. Scale bar: 10 µm. n=3 per group. (E,F) Immunofluorescence image of actin polymerisation (phalloidin, red) and nuclei (DAPI, blue) and protein levels of phosphorylated LATS1, total LATS1, phosphorylated YAP, total YAP in ADTC5 cells plated on TNC with or without administration of LPA for 12 hours. Scale bar: 10 µm. (G) Alcian blue staining of ADTC5 cells cultured on TNC transfected with empty vector or YAP-overexpressing vector for 7 days. (H) Protein levels of Sox9, Aggrecan and Collagen II in ADTC5 cells cultured on TNC transfected with empty vector or YAP-overexpressing vector for 48 hours. (I) Protein levels of YAP and Sox9 in ADTC5 cells plated on TNC with application of XMU-MP-1 or DMSO for 48 hours. (J,K) μCT images of new bone formation and quantitative analysis in DBA/1 mice with administration of XMU-MP-1 or DMSO for 16 weeks. Scale bar: 1 mm. n=9 per group. (L,M) H&E staining, SOFG staining, immunohistochemical analysis of Sox9 (indicated by red arrows) and quantitative analysis in DBA/1 mice with administration of XMU-MP-1 or DMSO for 8 weeks. Scale bar: 500 µm. n=5 per group. Data are presented as mean±SD. **p<0.01, *p<0.05, unpaired t-test. AS, ankylosing spondylitis; BV, bone volume; CB, cortical bone; DMSO, dimethyl sulfoxide; LPA, lysophosphatidic acid; N, non-AS patients; NB, new bone; SP, spinous process; TNC, tenascin-C; YAP, yes-associated protein.
To confirm that the chondrogenic effect of TNC was dependent on Hippo/YAP pathway activation and downstream phosphorylation and degradation of YAP, YAP was overexpressed in ADTC5 cells cultured under TNC-coated conditions. As expected, YAP overexpression significantly decreased the chondrogenesis induced by TNC (figure 5G,H, online supplemental figure S6D). Consistently, XMU-MP-1, a Hippo/YAP pathway antagonist that decreases the phosphorylation and degradation of YAP, also suppressed the expression of Sox9 and the chondrogenic differentiation of ADTC5 cells (figure 5I, online supplemental figure S6E,F).
To confirm that the activation of Hippo/YAP signalling is involved in pathological new bone formation in vivo, XMU-MP-1 was systemically administered to DBA/1 model. Pathological new bone was reduced in the XMU-MP-1-treated group, and the chondrogenic process was suppressed (figure 5J‒M). These results suggest that TNC-induced chondrogenesis depends on actin cytoskeleton depolymerisation-mediated YAP inactivation (online supplemental figure S6G,H).
TNC is majorly secreted by fibroblast-specific protein-1 (FSP1)+ fibroblasts
Single-cell RNA sequencing (scRNA-seq) of entheseal tissues from CAIA models was performed to explore candidates for TNC-secreting cells. Cluster analysis using t-distributed stochastic neighbour embedding dimensionality reduction identified 10 different cell clusters (figure 6A). TNC was majorly expressed in cluster 1 (fibroblasts) (figure 6B‒D). Consistent with the results of scRNA-seq, immunofluorescence staining showed that TNC was majorly co-stained with FSP1, which was a commonly used marker for fibroblasts in tissues from animal models (figure 6E) and patients with AS (figure 6F). Human fibroblasts isolated from ligament tissues were stimulated with different inflammatory cytokines related to AS pathogenesis, as identified in previous reports.3 5 The results showed that TNC was upregulated at both the mRNA and the protein levels by TNFα, IL-17A and IL-22 (online supplemental figure S7A‒C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TNC is majorly secreted by fibroblast-specific protein-1 (FSP1) +fibroblasts (A) T-distributed stochastic neighbour embedding (tSNE) plot of single-cell RNA sequencing from CAIA mice revealed 10 distinct cell clusters. (B) Violin plots of TNC. (C) Feature plot of TNC. (D) Feature plots displaying the single-cell gene expression of fibroblast, stem cells, inflammatory cells, chondrocytes and osteoblasts. (E) Double immunofluorescence staining in CAIA mice, including staining for FSP1, Sca-1, CD45, Sox9, MMP13, OCN and TNC. Scale bar: 100 µm. Semiquantitative analysis of TNC colocalisation. n=3. (F) H&E staining, SOFG staining and double immunofluorescence staining in spinal ligament tissues from patients with AS, including staining for FSP1, Sox9 and TNC. Scale bar: 200 µm. Semiquantitative analysis of TNC colocalisation. n=3. Data are presented as mean±SD. C, cartilage; CAIA, collagen antibody-induced arthritis; CB, cortical bone; L, ligament; NB, new bone; TNC, tenascin-C.
Discussion
Pathological new bone formation at the axial skeleton is one of the hallmark features of AS and causes spinal ankylosis, functional impairment and disability.3 Although numerous efforts have been made to explore the pathogenesis of this disease, the mechanisms underlying pathological new bone formation are not fully understood. In the current study, we identified a secreted matrix protein, TNC, that was aberrantly upregulated in ligament and entheseal tissues from patients with AS and animal models. In addition, we found that genetic ablation and pharmacological inhibition of TNC dramatically suppressed entheseal new bone formation, indicating its essential role in this pathological process.
Various types of bone formation, including endochondral ossification, membranous ossification and chondroid metaplasia, have been described in AS, among which endochondral ossification is considered to be the most important. During this process, new bone formation occurs after the formation of a cartilage template. Chondrocytes differentiate into hypertrophic chondrocytes, which are then replaced by osteoblasts to form mature bone.32 In the current study, we found that inhibiting TNC retarded the formation of the cartilage template, thereby suppressing subsequent pathological new bone formation. This finding was consistent with previous studies that have suggested the critical role of TNC in chondrogenesis and cartilage formation.14–16 The findings reveal that TNC-mediated cartilage formation is essential for subsequent pathological new bone formation.
ECM constructs the basic mechanical properties of the tissue microenvironment, including stress, strain, stiffness, elasticity and adhesion.34 It is accepted that the mechanical properties of tissues profoundly affect the differentiation process of mesenchymal stem cells, including osteogenesis, chondrogenesis and adipogenesis.35 36 The remodelling of ECM components is a pathological feature of chronically inflamed tissues.37 39 In this study, we found that aberrant TNC expression modified the ECM adhesion force and the subsequent mechanosignalling. TNC-mediated suppression of matrix adhesion force resulted in reduced nuclear localisation of YAP through the activation of Hippo pathway. Previous studies have shown that YAP is a negative regulator of chondrogenesis.40 41 Deng et al 42 showed that the dephosphorylation and nuclear localisation of YAP inhibited chondrocyte maturation by suppressing Col10a1 expression through interaction with Runx2.43 Similarly, Goto et al 44 found that dephosphorylation and nuclear localisation of YAP impaired chondrocyte proliferation and differentiation through the repression of Sox9. Consistently, we found that the dephosphorylation and nuclear localisation of YAP significantly inhibited chondrogenesis in vitro. Systemic administration of Hippo/YAP antagonist XMU-MP-1 led to a significant suppressive effect on entheseal cartilage formation and subsequent pathological new bone formation. Taken together, these results indicate that aberrant deposition of TNC in the entheseal microenvironment plays a vital role in influencing the mechanical properties of the matrix, resulting in YAP inactivation, and therefore enhancement of endochondral ossification.45 46
The relationship between inflammation and new bone formation in AS is still unclear.5 However, accumulating evidence shows that inflammation is directly involved in the pathological process of new bone formation, including enhancement of osteoinductive protein production and promotion of osteoprogenitor proliferation.47 48 In this study, we further propose that inflammation potentiates new bone formation by remodelling the ECM. The results of immunohistochemical staining showed infiltration of inflammatory cells and expression of inflammatory cytokines in the tissue samples collected from patients with AS, indicating that the regions of potential pathological new bone are inflamed, which is consistent with previous studies.49–51 scRNA-seq analysis and immunofluorescence staining revealed that TNC was primarily secreted by FSP1+fibroblasts. FSP1, also known as S100A4, is a widely reported fibroblast marker. FSP1 is mainly expressed in fibroblasts of various organs undergoing tissue remodelling.52 53 Fibroblasts are the majority of cells in enthesis/ligament tissues and largely proliferate on inflammation stimulation.24 In addition, fibroblasts are also well acknowledged as secretary cells that produce various ECM proteins or cytokines to participate in the regulation of the microenvironment during multiple pathological processes.52 In AS, a previous study showed that fibroblast-rich granulation tissue promotes new bone formation.54 In an in vitro study, we confirmed that fibroblasts produced large amounts of TNC under the stimulation of various AS-associated inflammatory cytokines, including TNFα, IL-17A and IL-22. Taken together, it suggests that inflammation-induced aberrant TNC expression and TNC-mediated ECM remodelling contribute to the formation of an osteoinductive microenvironment and potentiate new bone formation via alteration of tissue mechanical cues. In addition, the fact that multiple inflammatory cytokines can induce TNC production explains, to a certain extent, the low pharmacological efficacy on the radiographic progression of patients with AS of antibodies that neutralise a single cytokine, such as adalimumab, ustekinumab and risankizumab.47 48
TNC is as an intriguingly multifunctional molecule that exhibits diverse roles in immunity, such as in the promotion of bacterial adhesion and thrombosis, in the regulation of innate and adaptive immunity, and in the control of ECM synthesis and remodelling during tissue repair.55 Normally, TNC expression is precisely regulated. During physiologic responses to injuries or infection, it is induced at sites of inflammation and peaks once tissue rebuilding commences and down-regulates concomitant with the resolution of inflammation and tissue repair.55 On the contrary, during abnormal wound-healing responses or pathologies associated with persistent inflammation, prolonged expression of TNC is observed. Abnormal regulation of TNC expression is found responsible for the long-lasting inflammation and pathological rebuilding in many diseases.11 55 In the studies of rheumatoid arthritis (RA), TNC has been demonstrated as an endogenous activator of Toll-like receptor 4, which is responsible for maintaining inflammation and joint destruction.18 56 TNC has also been shown to exhibit proinflammatory effects by activating α9 integrins in macrophages, resulting in the production of various proinflammatory molecules in the development of arthritis.57 In addition, post-translationally citrullinated TNC achieved increased immunogenicity of the C-terminal residues, leading to the generation of autoantibodies in patients with RA.58 Recently, serum level of TNC was reported to be elevated in patients with AS and associated with disease activity,19 but which role that TNC plays in AS was unclear. In the current study, we found that TNC was involved in pathological new bone formation in AS, which could be considered as a special form of abnormal tissue remodelling. However, although our results provide evidence that TNC contributed to pathological new bone formation through modulation of the biomechanical property of ECM and enhancement of chondrogenesis, given that it plays multifaceted roles in immunomodulation and inflammation, TNC might also have other critical roles in the regulation of entheseal and ligamentous microenvironment or the maintenance of chronic inflammation in AS. It will be of great interest to continue to investigate the contribution of this multifunctional molecule to the pathogenesis of AS.
There are some limitations of the current study. First, control samples from healthy individuals are extremely difficult to obtain. Nearly all of the age-matched and sex-matched controls in our research were patients suffering from adult idiopathic scoliosis. Although the sample collection location was far from the apical vertebra and spontaneous fusion zone, gene expression in these samples may still be different from that in undamaged healthy human tissue samples. Second, the AS tissue samples were collected from patients at late-stage with extensive spinal fusion. Whether the biomechanical changes of the spine contribute to the pathological process of new bone formation is unclear due to lack of available tissue samples from patients at early-stage as proper control, which requires further investigation. Third, although the AS animal models in the current study are well accepted, the triggers in rodent models may not be identical to those in human disease. For further development of therapeutic strategies, large animal models whose genetic background is more similar to that of human, will likely be required.
In summary, we demonstrated that exposure of entheseal sites to chronic inflammation causes excessive TNC deposition, which subsequently promotes chondrogenic differentiation and pathological new bone formation via suppression of ECM adhesion force and activation of the Hippo pathway. Suppression of aberrant expression of TNC may be a potential therapeutic strategy for pathological new bone formation in AS.
Data availability statement
Data are available upon reasonable request. The data that support the findings in this study are available from the corresponding author upon request.
Ethics statements
Ethics approval
The Medical Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University approved the procedures performed in this study.
Acknowledgments
We gratefully acknowledge technical sequencing and analysis from GENE DENOVO and knockout mice from Cyagen Biosciences.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
ZihaoL and SC contributed equally.
Contributors HL conceived the ideas for experimental designs. ZihaoL and SC conducted the majority of the experiments, analysed data and prepared the manuscript. DC and ZhongpingZ conducted sample collection and performed statistical analysis. JW, ZeminL and ZhaominZ provided critical suggestions and instructions for the project and helped compose the manuscript. ZihaoL and SC provided μCT analysis. ZihaoL, SC, XL, HC and WH conducted most animal experiments and performed analysis. HL developed the concept, supervised the project and conducted data analysis.
Funding The work was supported by National Natural Science Foundation of China (Grant no 81972039; 81772307; 81572103), Science and Technology Planning Project of Guangdong Province, China (Grant no 2017A050501016), Special Support Plan for High-Level Talent of Guangdong Province, China (Grant no 2016TQ03R667), Pearl River Nova Program of Guangzhou, China (Grant no 201610010103) and KELIN New Talent Project of The First Affiliated Hospital, Sun Yat-sen University (Grant no Y12001).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.