Article Text

Abstract
Objective The greatest genetic effect reported for systemic sclerosis (SSc) lies in the major histocompatibility complex (MHC) locus. Leveraging the largest SSc genome-wide association study, we aimed to fine-map this region to identify novel human leucocyte antigen (HLA) genetic variants associated with SSc susceptibility and its main clinical and serological subtypes.
Methods 9095 patients with SSc and 17 584 controls genome-wide genotyped were used to impute and test single-nucleotide polymorphisms (SNPs) across the MHC, classical HLA alleles and their composite amino acid residues. Additionally, patients were stratified according to their clinical and serological status, namely, limited cutaneous systemic sclerosis (lcSSc), diffuse cutaneous systemic sclerosis (dcSSc), anticentromere (ACA), antitopoisomerase (ATA) and anti-RNApolIII autoantibodies (ARA).
Results Sequential conditional analyses showed nine SNPs, nine classical alleles and seven amino acids that modelled the observed associations with SSc. This confirmed previously reported associations with HLA-DRB1*11:04 and HLA-DPB1*13:01, and revealed a novel association of HLA-B*08:01. Stratified analyses showed specific associations of HLA-DQA1*02:01 with lcSSc, and an exclusive association of HLA-DQA1*05:01 with dcSSc. Similarly, private associations were detected in HLA-DRB1*08:01 and confirmed the previously reported association of HLA-DRB1*07:01 with ACA-positive patients, as opposed to the HLA-DPA1*02:01 and HLA-DQB1*03:01 alleles associated with ATA presentation.
Conclusions This study confirms the contribution of HLA class II and reveals a novel association of HLA class I with SSc, suggesting novel pathways of disease pathogenesis. Furthermore, we describe specific HLA associations with SSc clinical and serological subtypes that could serve as biomarkers of disease severity and progression.
- systemic sclerosis
- autoantibodies
- immune complex diseases
- polymorphism
- genetic
Data availability statement
Summary statistics are available from the corresponding author upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
The major histocompatibility complex is the genomic region shown to have the greatest genetic effect in several autoimmune diseases such as systemic sclerosis (SSc).
What does this study add?
Taking advantage of the largest genetic study in SSc, we conducted an extensive fine-mapping of the region by assessing single nucleotide polymorphisms, human leucocyte antigen (HLA) classical alleles and polymorphic amino acid residues associated with SSc.
We have confirmed the strong contribution of HLA class II in SSc susceptibility and showed for the first time the independent association of HLA class I, suggesting novel pathways of disease pathogenesis.
We have identified specific associations in the different clinical forms of the disease, as well as private associations regarding autoantibodies presentation.
How might this impact on clinical practice or future developments?
These findings may improve our knowledge of disease onset and progression, as well as assist in the identification of biomarkers that allow early and specific interventions.
Introduction
Genetic variation within the major histocompatibility complex (MHC) has been associated with many human conditions, particularly autoimmune and infectious diseases or those with a central immunological component.1 2 Systemic sclerosis (SSc) or scleroderma is a rare systemic immune-mediated inflammatory disease (IMID), with a broad spectrum of clinical forms, affecting primarily connective tissues.3 It is characterised by an immunological disturbance leading to the production of autoantibodies, vascular damage, and widespread fibrosis of the skin and internal organs.3–5 Regarding the clinical characteristics of the disease, patients with SSc are classified depending on the extent of the dermal fibrosis as either limited cutaneous systemic sclerosis (lcSSc) and diffuse cutaneous systemic sclerosis (dcSSc).3 5 6 Further classifications are performed according to the immunological dysregulation and the mutually exclusive production of autoantibodies in anticentromere (ACA), antitopoisomerase (ATA) and anti-RNA polymerase III (ARA) antibodies.3 6
SSc is a complex disease, in which the contributions of environmental and genetic factors are crucial for disease onset and progression.7–9 Several genome-wide association studies (GWASs) and Immunochip analyses have shed light into this genetic component.10–12 Interestingly, a recent GWAS in SSc confirmed that the greatest genetic contribution to the disease described thus far comes from the human leucocyte antigen (HLA) region.13 Genetic variations in the HLA system may determine their binding affinity for specific antigens and their presentation to antigen-presenting cells, leading to the activation of autoreactive T-helper and B cells and the production of autoantibodies.14 These genetic variations are detected in seropositive IMIDs, and several of them have been described to be shared among them.15 16 HLA fine-mapping studies have been carried out successfully in several IMIDs, including rheumatoid arthritis (RA),17 systemic lupus erythematosus (SLE)18 and myositis,19 among others, and have been proven useful in identifying the strongest genetic risk factors in autoimmune diseases.2 In SSc previous assessments identified polymorphic amino acid positions and single-nucleotide polymorphisms (SNPs) that modelled the observed associations in populations of European descent,11 12 and a recent study in African and European confirmed an African ancestry-predominant allele and a transancestry association with individuals of European ancestry.20 21
Taking the aforementioned into consideration and leveraging the enhanced power provided by the most recent GWAS in SSc, we conducted a broad analysis of the MHC region to evaluate SNPs, classical HLA alleles and their polymorphic amino acid positions, with SSc and its clinical and serological subphenotypes. We also functionally explored the associated variants, finding evidence of colocalisation with expression quantitative trait loci (eQTLs).
Materials and methods
Study population
This study included genome-wide genotyped data from 9846 patients with SSc and 18 333 healthy individuals from the same source population.13 The patients fulfilled the 2013 American College of Rheumatology/the European League Against Rheumatism classification criteria or the criteria proposed by LeRoy and Medsger for early SSc.22 23 In addition, patients were stratified by the main clinical classifications (lcSSc or dcSSc) and main autoantibody status (ACA, ATA or ARA). Details of the cohorts, genotyping methods and quality control (QC) for genotyped data are described elsewhere.13
SNP and HLA imputation
After genotyping QC, SNPs, classical HLA alleles and amino acid variants, were all imputed for each case–control dataset separately in the extended MHC region in chromosome 6.24 The SNP2HLA25 software was used for imputation using a reference panel consisting of 5225 European individuals in the Type 1 Diabetes Genetic Consortium,26 containing data of 8961 variants across the MHC region, and two and four digit-resolution allelic identities of the HLA class I (HLA-A, HLA-B and HLA-C) and II genes (HLA-DPA1, HLA-DPB1, HLA-DQA1, HLA-DQB1 and HLA-DRB1) as well as their amino acid make-up. Imputed data were also filtered for 95% success call rate for alleles and amino acids, deviation from Hardy-Weinberg equilibrium (HWE) considering a p-value of <0.001 for SNPs in controls and 95% total call rate for individuals. The total numbers of imputed variants per case–control set are specified in online supplemental table 1.
Supplemental material
Supplemental material
Statistical analysis
Association analyses were performed with PLINK27 using logistic regressions in each of the 14 independent datasets, including sex and the five first principal components (PCs) as covariates. Briefly, PC analysis was performed using ~1 00 000 quality-filtered independent SNPs outside the MHC region using PLINK and GCTA64. Outliers were identified and removed as described elsewhere.13 We tested SNPs, classical HLA alleles and all possible combinations of amino acid residues per position. Inverse variance fixed effects meta-analysis was conducted with PLINK to evaluate the consistency of effects across studies. The genome-wide significance was established at a p-value ≤5×10−8.
Considering the main clinical SSc subtypes and serological classifications, stratification of cases was performed following the same procedure as for the global analysis and comparisons were made with the control group and intracases, namely, dcSSc with lcSSc, and ATA with ACA (patients without available data or positive for both autoantibodies were excluded from the analysis). Only classical alleles whose results outperformed those from the global analysis and that were significantly associated in both comparisons were declared as private.
To identify independent signals within the region, sequential conditional association analyses were performed with the software GCTA-COJO19 28 29 using the summary statistics from the meta-analysis (global and stratified by clinical and serological subtypes) and separately for each variant type (SNPs, alleles,and amino acids). The Manhattan plot was obtained with an in-house R script. The Protein Data Bank entries 3pdo, 1a1m, 3lqz and 2bvp were used to produce the 3D models of the HLA molecules with the UCSF Chimaera software.30
Functional assessment of associated variants
In order to assign a biological meaning of our association results at the SNP level, we performed a colocalisation analysis using COLOC31 and the Genotype-Tissue Expression (GTEx) project release V.8 (dbGaP Accession phs000424.v8.p2). Colocalisation analysis evaluates if two independent studies at the same locus consistently share a causal variant; if so, the probability of a causal association increases.
Results
A total of 9095 patients with SSc and 17 584 healthy individuals fulfilled the QCs and 8339 variants were meta-analysed, including SNPs, classical alleles and amino acid positions within the MHC region (online supplemental table 1), identifying 1273 reaching the genome-wide level of significance (figure 1).

Association signals for systemic sclerosis in the human leucocyte antigen region. The −log10 of the meta-analysis p values are plotted against their chromosomal position. The red line represents the genome-wide level of significance (p value=5×10−08). The size of the diamond indicates the degree of linkage disequilibrium with the strongest association from the meta-analysis (rs1048372).
SNP and HLA associations
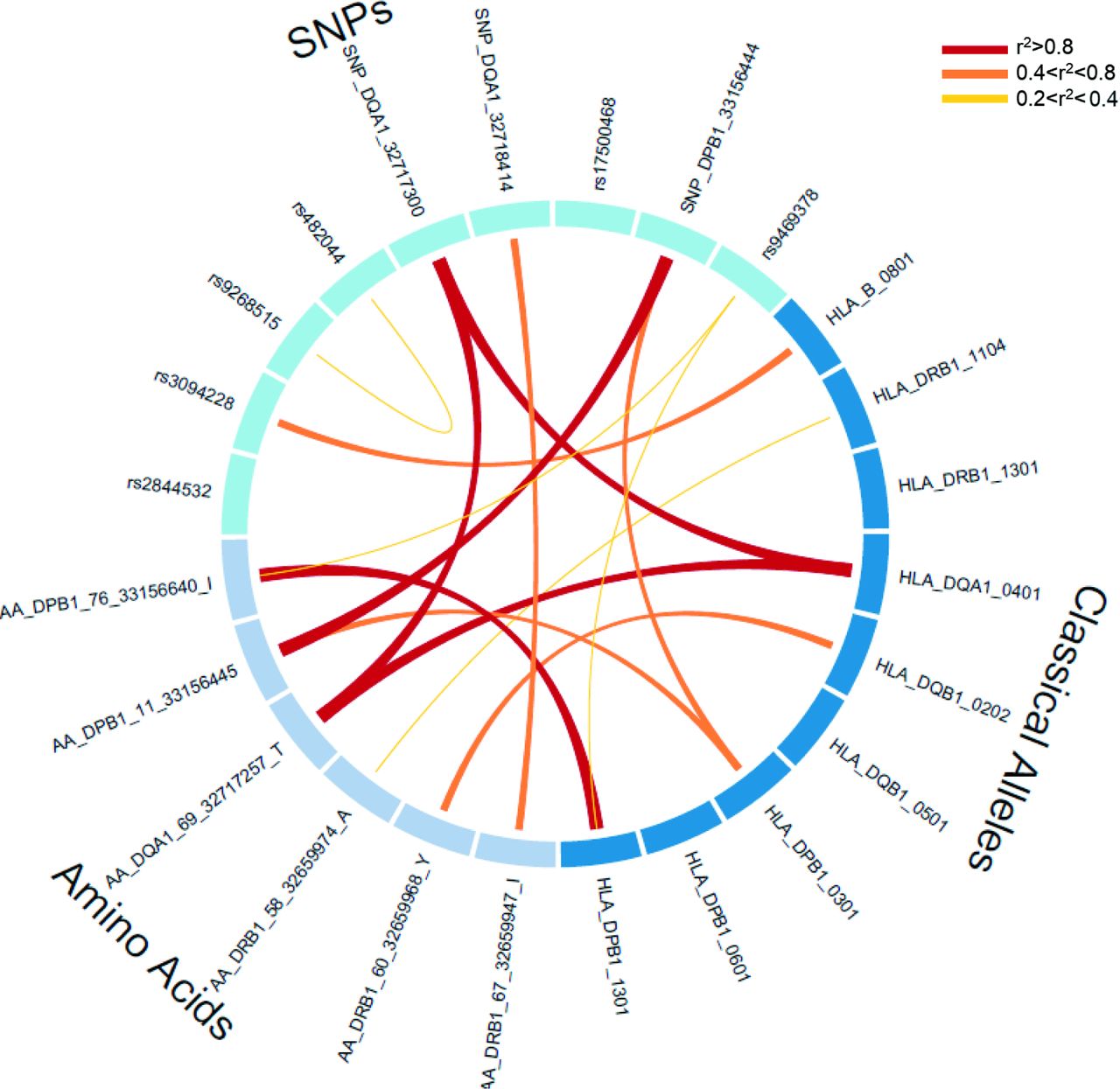
Within this region, the global meta-analysis yielded 1082 significantly associated SNPs, from which nine were independent and modelled the observed SNPs associations in the region (table 1) after the sequential conditional analysis. The most associated signal corresponded to a protective synonymous coding SNP in the HLA-DQA1 gene (rs1048372, OR=0.70, 95% CI 0.67 to 0.73, p value=1.29×10−63). In addition, another synonymous coding SNP in the same gene was independently associated with SSc (rs1142338, OR=1.86, 95% CI 1.67 to 2.07, p value=3.16×10−12) and a truncating SNP mapping in the HLA-DPB1 gene (rs1126511, OR=1.21, 95% CI 1.16 to 1.27, p value=2.37×10−25). All the remaining SNPs were non-coding intronic/intergenic, potentially involved in the regulation of gene expression. Given the complex linkage disequilibrium (LD) structure in the region, we assessed the relationship among the associated variants, and rs2844532 and rs17500468 were not in LD with any classical alleles and amino acid residues (figure 2).
Independent association results from the global analysis comparing scleroderma and controls after the sequential conditional analysis
{kind=link}
{kind=link}
LD among the independent variants. Circos plot depicting the LD relationship among the SNPs, four-digit classical HLA alleles and HLA amino acid residues independently associated from the sequential conditional analysis. HLA, human leucocyte antigen; LD, linkage disequilibrium; SNP, single-nucleotide polymorphism.
In the global meta-analysis and regarding the classical alleles, a total of 21 four-digit classical alleles were significantly associated with SSc at the genome-wide level with strong signals within the HLA class II. The strongest association observed was with HLA-DRB1*11:04 (OR=2.11, 95% CI 1.92 to 2.31, p value=2.52×10−56). After sequential conditional analysis controlling for the effect of the most associated HLA alleles, seven additional class II genes were independently associated, including three HLA-DPB1, two HLA-DQB1, one HLA-DQA1 and one HLA-DRB1 alleles (table 1). Interestingly, here we describe for the first time the independent association with SSc of an HLA class I conferring risk, which belongs to the classical allele HLA-B*08:01 (OR=1.22, 95% CI 1.15 to 1.30, p value=1.79×10−12) (table 1). The HLA-DRB1*13:01, HLA-DQB1*05:01 and the HLA-DPB1*06:01 were independent (r2<0.2) from the other variants irrespective of their nature (amino acid residues and SNPs) (figure 2).
Moreover, we performed amino acid analysis with a total of 170 polymorphic amino acid residues significantly associated in the global SSc meta-analysis. The most significant amino acid residue associated with SSc was the Ile67 of the HLA-DRβ1 (OR=0.70, 95% CI 0.67 to 0.73, p value=1.70×10−63). A summary of the independent associations after the stepwise conditional model is shown in table 1 and online supplemental figure S1A–C. All the associated amino acid residues were in moderate to high LD (0.4<r2<0.8) with the reported classical alleles and SNPs (online supplemental table 2).
Supplemental material
Functional annotation of associated SNPs
To functionally characterise the associations from the meta-analysis and their proxies (r2≥0.8) at the SNP level, they were tested against the eQTLs from the 49 tissues contained in GTEx by a colocalisation approach.31 We identified 70 SNPs affecting the expression of 82 eGenes with a posterior probability of 80% in 40 tissues (online supplemental table 3). Then, we further assessed their overlap with the independent variants or any proxies and identified five of them affecting the expression of 11 eGenes in relevant cells and tissues involved in the disease, including lymphocytes, fibroblasts, colon and oesophagus, among others (table 2).
Colocalisation analysis for the independently associated SNPs
Clinically restricted subphenotype analysis
The numbers of patients in each subgroup are summarised in online supplemental table 4. Given that previous studies reported genetic differential susceptibility to SSc depending on its subtype,12 13 32 we performed stratified analyses comparing with the control group and the results are summarised in the online supplemental tables 5‒9. For lcSSc, a total of six classical alleles were identified as independently associated (table 3 and online supplemental table 5). Among them, HLA-DQA1*02:01 was only associated with lcSSc (OR=0.54, p value=5.23×10−51), and this was further confirmed when compared with the patients with dcSSc (OR=0.71, p value=2.08×10−8; online supplemental table 10). Regarding dcSSc, four classical alleles were independently associated with this subphenotype when compared with the healthy individuals (table 3 and online supplemental table 6). HLA-DQA1*05:01 was exclusively associated with dcSSc (OR=1.49, p value=1.59×10−11). This was confirmed when comparing these patients with patients with lcSSc (OR=1.30, p value=1.76×10−11) (online supplemental table 10).
Summary of the independent association results from the stratified analysis
Serologically restricted analysis
Taking into account the serologically restricted subphenotypes, we conducted different analyses to compare ACA, ATA and ARA positive cases and controls in this locus. In the case of ACA, HLA-DRB1*08:01 (OR=3.18, p value=4.00×10−64) and HLA-DRB1*07:01 (OR=0.36, p value=1.84×10−45) were the classical alleles independently and exclusively associated with the presence of this autoantibody (online supplemental table 7), and this was verified when comparing with the ATA-positive patients (OR=2.17, p value=1.42×10−10 and OR=0.42, p value=3.85×10−27, respectively) (table 3 and online supplemental table 10). Concerning the analysis in the ATA subgroup, two classical alleles, namely, HLA-DPA1*02:01 and HLA-DQB1*03:01 were significantly and exclusively associated with this phenotype (OR=1.87, p value=2.93×10−19 and OR=1.86, p value=7.00×10−19, respectively) (online supplemental table 8), which was confirmed in the intracases comparison (OR=2.41, p value=1.09×10−40 and OR=1.67, p value=1.73×10−22, respectively) (table 3). Regarding the ARA-positive analysis, only HLA-DRB1*11:04 was significantly associated with the presence of this autoantibody (online supplemental table 9).
Given the known correlation between the subphenotypes and the autoantibodies,3 it is worth noting the overlap of HLA-DRB1*08:01 as associated with lcSSc (OR=2.18, p value=8.07×10−29) and with ACA-positive patients (OR=3.18, p value=4.00×10−64); however, this association was no longer significant for lcSSc when compared with dcSSc (OR=1.49, p value=3.43×10−5) (online supplemental tables 5 and 7).
Discussion
Leveraging the largest genetic study conducted in SSc, we performed a comprehensive analysis of the MHC locus by fine-mapping approaches involving SNPs and imputed four-digit classical HLA alleles and their amino acid residues. Our results showed strong evidence for the substantial contribution of the HLA class II region in the pathophysiology of SSc, with strong associations with HLA-DRB1*11:04, HLA-DQB1*02:02 and HLA-DPB1*13:01 alleles. Furthermore, we revealed for the first time the genome-wide significant association of a class I HLA gene, the HLA-B*08:01. In addition, we identified associated amino acid residues in several HLA type II genes, mapping in the peptide binding pocket and non-coding variants involved in gene expression regulation. Importantly, the stratified analysis showed HLA-DQA1*02:01 as associated with the lcSSc subtype, in contrast to HLA-DQA1*05:01 that was private for dcSSc. Likewise, the serological stratification also showed exclusive associations; for instance, HLA-DRB1*08:01 and HLA-DRB1*07:01 alleles were significantly associated in ACA-positive patients unlike in the ATA-positive patients, where associations with the HLA-DPA1*02:01 and HLA-DQB1*03:01 alleles were detected. In ARA-positive patients only HLA-DRB1*11:04 was significantly associated with this autoantibody presentation.
In the global analysis, three of the most associated alleles in the model, that is, HLA-DRB1*11:04, HLA-DQB1*02:02 and HLA-DPB1*13:01, have been previously reported as associated with SSc in different populations,21 33–36 confirming them as robustly associated with the disease. In addition, the risk allele rs17500468*G, which is an intronic variant mapping in the HLA-DQA2 gene, was previously reported in an Immunochip study11 and is in tight LD (D’=1.0) with rs2857130 identified in a further Immunochip,12 validating its association in European population.11 The current study revealed the genome-wide significant association of HLA-B*08:01. Other autoimmune diseases such as RA, SLE, myositis, Sjögren’s syndrome (SjS) and primary sclerosing cholangitis17 37 38 with strong HLA class II associations, have also shown HLA class I associations, as described here for SSc. A haplotypic block containing this allele was previously nominally associated in Mexican patients with SSc.35 These HLA-B*08 associations have been attributed to the long ancestral 8.1 haplotype, supporting a common genetic background in autoimmunity.16 39–41 This allele is in high LD (r2=0.998) with the amino acid residue Asp9 located in the peptide binding groove of HLA-B, with a potential functional impact on antigen presentation.17
The association with SSc of independent signals in HLA classes I and II may suggest novel mechanisms for disease pathogenesis, including the involvement of not only CD4+ but also CD8+ T cells.2 42 Interestingly, genes associated with CD8+ T-cell biology have been reported to be deregulated in skin biopsies of active SSc lesions, and these cells have been described to produce proinflammatory cytokines, contributing to the overproduction of collagen by fibroblasts and excessive fibrosis.43 A recent report by Maehara et al assessed T-cell infiltrates in the skin of early dcSSc and showed that CD4+ cytotoxic T cells and CD8+ T cells are responsible for these infiltrates and induce apoptotic death of endothelial cells, contributing to the vasculopathy and fibrotic environment observed in SSc.44 45 Functional maturation defects have been detected in regulatory CD8+ lymphocytes from an ex vivo model of SSc46 and differential regulatory programmes of IFN-associated genes in CD4+ and CD8+ T cells have been shown to lead to elevated serum interferon levels in patients with SSc .47 Taken altogether, further studies on the contribution of CD8+ T cells in SSc may bear great therapeutic value, due to either its connection with the development of fibrosis or the assessment of the subpopulation of regulatory CD8+ cells in these patients.
Given that most of the independent SNPs in the global analysis mapped in non-coding regions of the genome, we performed a colocalisation study to assess if the associated variants were modulating gene expression. Our data showed immunity-related genes such as HLA-DRB1, HLA-DRB6, HLA-DPA2 and the complement gene C2 as eGENES regulated by the associated variants (table 2). Specifically, the risk allele of rs4713586 is correlated with an increased expression of the C2 gene in transformed lymphocytes. This gene was previously associated with SLE48 and psoriasis.49 Interestingly, genetic variations on the complement genes have been recently described to contribute to the sex-biased susceptibility in highly related diseases like SLE and SjS,50 and may be further explored in SSc. This could be seen as a limitation of our study because the reference panel used here does not allow the imputation of these structural variations.
One potential application of genetic studies is the identification of variants associated with clinical and serological subtypes to assist in patient stratification, and potentially to anticipate their progression and to propose specific therapeutic interventions. The determination of classical HLA alleles is routine in immunology laboratories for autoimmune diseases such as coeliac disease, ankylosing spondylitis and type 1 diabetes, and could be extended to others like SSc. To this aim, our stratified analysis showed that HLA-DQA1*02:01 was significantly associated with lcSSc, while HLA-DQA1*05:01 was exclusively associated with dcSSc. Regarding the serological stratifications, HLA-DRB1*08:01 and HLA-DRB1*07:01 were associated with ACA positive patients, further confirming associations reported in previous candidate gene GWAS, and Immunochip studies.11 12 51 In the ATA-positive SSc subgroup, HLA-DPA1*02:01 and HLA-DQB1*03:01 showed exclusive and significant associations, and the latter was also reported in an Immunochip study.11
It is worth noting that the private associations were stronger when stratifying by the clinical and serological group of patients, despite the considerable loss of statistical power (online supplemental table 10). These results highlight the importance of analysing homogeneous groups of patients, reducing the loss of power due to phenotypical heterogeneity.52 As expected, these alleles were significantly different even when comparing the group of patients among them and not with the control group, reinforcing the idea that they are present in specific clinical and serological subtypes of patients. Overall, the risk alleles identified thus far bear modest effects and a better understanding of the genetic structure of the disease will include interactions between several risk factors. Further studies warrant the simultaneous qualitative and quantitative assessments of allele-specific expression of the genes in order to detect context-specific regulatory effects.53 54 Genotyping equivalent SNPs to the associated HLA alleles may also be of clinical utility, as SNP genotyping is straightforward and cost-efficient and has been proven to be very valuable to infer classical alleles for this and other rheumatic diseases.21 55 56
In summary, our extensive study of the HLA genes has confirmed and revealed novel associations with SSc susceptibility, highlighting for the first time the involvement of HLA class I genes in the pathogenesis of the disease. In addition, our data points to specific allelic associations that may serve as molecular biomarkers of clinical disease and serological subphenotypes. This evidence may eventually lead to early interventions that are crucial to avoid the devastating effects of the disease, and to develop specific and effective therapeutic options for patients with SSc.
Data availability statement
Summary statistics are available from the corresponding author upon reasonable request.
Ethics statements
Ethics approval
CSIC’s ethics committee approved the study protocol, and written informed consent was obtained in accordance with the tenets of the Declaration of Helsinki. This study was conducted using available data included in a previously published genome-wide association study.
Acknowledgments
We thank Sofia Vargas, Sonia García and Gema Robledo for their excellent technical assistance and all the patients and control donors for their essential collaboration. We also thank National DNA Bank Carlos III (University of Salamanca, Spain), who supplied part of the control DNA samples from Spain, WTCCC and EIRA Consortiums, and PopGen 2.0 network.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Twitter @ShervinAssassi
Collaborators International SSc Group: P Carreira, Department of Rheumatology, 12 de Octubre University Hospital, Madrid, Spain; I Castellvi, Department of Rheumatology, Santa Creu i Sant Pau University Hospital, Barcelona, Spain; R Ríos, Department of Internal Medicine, San Cecilio Clinic University Hospital, Granada, Spain; J L Callejas, Department of Internal Medicine, San Cecilio Clinic University Hospital, Granada, Spain; R García Portales, Department of Rheumatology, Virgen de la Victoria Hospital, Málaga, Spain; A Fernández-Nebro, Department of Rheumatology, Carlos Haya Hospital, Málaga, Spain; F J García-Hernández, Department of Internal Medicine, Virgen del Rocío Hospital, Sevilla, Spain; M A Aguirre, Department of Rheumatology, Reina Sofía/IMIBIC Hospital, Córdoba, Spain; B Fernández-Gutiérrez, Department of Rheumatology, San Carlos Clinic Hospital, Madrid, Spain; L Rodríguez-Rodríguez, Department of Rheumatology, San Carlos Clinic Hospital, Madrid, Spain; P García de la Peña, Department of Rheumatology, Madrid Norte Sanchinarro Hospital, Madrid, Spain; E Vicente, Department of Rheumatology, La Princesa Hospital, Madrid, Spain; J L Andreu, Department of Rheumatology, Puerta de Hierro Hospital-Majadahonda, Madrid, Spain; M Fernández de Castro, Department of Rheumatology, Puerta de Hierro Hospital-Majadahonda, Madrid, Spain; F J López-Longo, Department of Rheumatology, Gregorio Marañón University Hospital, Madrid, Spain; V Fonollosa, Department of Internal Medicine, Valle de Hebrón Hospital, Barcelona, Spain; A Guillén, Department of Internal Medicine, Valle de Hebrón Hospital, Barcelona, Spain; G Espinosa, Department of Internal Medicine, Clinic Hospital, Barcelona, Spain; C Tolosa, Department of Internal Medicine, Parc Tauli Hospital, Sabadell, Spain; A Pros, Department of Rheumatology, Hospital Del Mar, Barcelona, Spain; E Beltrán, Department of Rheumatology, Hospital Del Mar, Barcelona, Spain; M Rodríguez Carballeira, Department of Internal Medicine, Hospital Universitari Mútua Terrasa, Barcelona, Spain; F J Narváez, Department of Rheumatology, Bellvitge University Hospital, Barcelona, Spain; M Rubio Rivas, Department of Internal Medicine, Bellvitge University Hospital, Barcelona, Spain; V Ortiz-Santamaría, Department of Rheumatology, Granollers Hospital, Granollers, Spain; A B Madroñero, Department of Internal Medicine, Hospital General San Jorge, Huesca, Spain; M A González-Gay, Epidemiology, Genetics and Atherosclerosis Research Group on Systemic Inflammatory Diseases, DIVAL, University of Cantabria, Santander, Spain; B Díaz, Department of Internal Medicine, Hospital Central de Asturias, Oviedo, Spain; L Trapiella, Department of Internal Medicine, Hospital Central de Asturias, Oviedo, Spain; M V Egurbide, Department of Internal Medicine, Hospital Universitario Cruces, Barakaldo, Spain; P Fanlo-Mateo, Department of Internal Medicine, Hospital Virgen del Camino, Pamplona, Spain; L Saez-Comet, Department of Internal Medicine, Hospital Universitario Miguel Servet, Zaragoza, Spain; F Díaz, Department of Rheumatology, Hospital Universitario de Canarias, Tenerife, Spain; J A Roman-Ivorra, Department of Rheumatology, Hospital Universitari i Politecnic La Fe, Valencia, Spain; J J Alegre Sancho, Department of Rheumatology, Hospital Universitari Doctor Peset, Valencia, Spain; M Freire, Department of Internal Medicine, Thrombosis and Vasculitis Unit, Complexo Hospitalario Universitario de Vigo, Vigo, Spain; F J Blanco Garcia, Department of Rheumatology, INIBIC-Hospital Universitario A Coruña, La Coruña, Spain; N Oreiro, Department of Rheumatology, INIBIC-Hospital Universitario A Coruña, La Coruña, Spain; T Witte, Department of Clinical Immunology, Hannover Medical School, Hannover, Germany; A Kreuter, Department of Dermatology, Josefs-Hospital, Ruhr University Bochum, Bochum, Germany; G Riemekasten, Clinic of Rheumatology, University of Lübeck, Lübeck, Germany; P Airo, Service of Rheumatology and Clinic Immunology Spedali Civili, Brescia, Italy; C Magro, Department of Rheumatology, Leiden University Medical Center, Leiden, The Netherlands; A E Voskuyl, Department of Rheumatology, VU University Medical Center, Amsterdam, The Netherlands; M C Vonk, Department of Rheumatology, Radboud University Nijmegen Medical Center, Nijmegen, Netherlands; R Hesselstrand, Department of Rheumatology, Lund University, Lund, Sweden; A Nordin, Division of Rheumatology, Department of Medicine, Karolinska University Hospital, Karolinska Institute, Stockholm, Sweden; C Lunardi, Department of Medicine, Università degli Studi di Verona, Verona, Italy; A Gabrielli, Istituto di Clinica Medica Generale, Ematologia ed Immunologia Clinica, Università Politecnica delle Marche, Ancona, Italy; A Hoffmann-Vold, Department of Rheumatology, Oslo University Hospital, Oslo, Norway; J H W Distler, Department of Internal Medicine 3, Institute for Clinical Immunology, University of Erlangen-Nuremberg, Erlangen, Germany; L Padyukov, Division of Rheumatology, Department of Medicine, Karolinska University Hospital, Karolinska Institute, Stockholm, Sweden; B P C Koeleman, University Medical Center Utrecht, Utrecht, The Netherlands. Australian Scleroderma Interest Group: W Stevens, St. Vincent’s Hospital, Melbourne, Victoria, Australia; M Nikpour, The University of Melbourne at St. Vincent’s Hospital, Melbourne, Victoria, Australia; J Zochling, Menzies Research Institute Tasmania, University of Tasmania, Hobart, TAS, Australia; J Sahhar, Department Rheumatology, Monash Medical Centre, Melbourne, VIC, Australia; J Roddy, Rheumatology, Royal Perth Hospital, Perth, WA, Australia; P Nash, Research Unit, Sunshine Coast Rheumatology, Maroochydore, QLD, Australia; K Tymms, Canberra Rheumatology, Canberra, ACT, Australia; M Rischmueller, Department Rheumatology, The Queen Elizabeth Hospital,Woodville, SA, Australia; S Lester, Department Rheumatology, The Queen Elizabeth Hospital, Woodville, SA, Australia.
Contributors Data analysis, manuscript drafting, revision and approval: MA-H; interpretation of data, manuscript revision and approval: MK and EL-I; sample and data collection: SA, LB, CPS-A, NO-C, ISG, SMP, ASIG, NH, GM, JkdV-B, GO, AB, ALH, CT, YA, MAB, TRDJR, CF, CPD and MDM; Study design, manuscript drafting, revision and approval: JM. All coauthors revised the work critically and made substantial contributions for important intellectual content.
Funding This work was supported by the Spanish Ministry of Science and Innovation (grant ref. SAF2015-66761-P and RTI20181013 (32-B-100)), Red de Investigación en Inflamación y Enfermedades Reumáticas from Instituto de Salud Carlos III (RD16/0012/0013) and grants from National Institutes of Health (R01AR073284) and DoD (W81XWH-16-1-0296). MAH was funded by the Spanish Ministry of Science and Innovation through the Juan de la Cierva Incorporacion program (ref. IJC2018-035131-I). GO, AB and ALH were supported by the NIHR Manchester Biomedical Research Centre and Versus Arthritis (grant ref 21754).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.