Article Text

Statistics from Altmetric.com
- AIA, adjuvant induced arthritis
- Ang, angiopoietin
- bFGF, basic fibroblast growth factor
- CIA, collagen induced arthritis
- COX, cyclo-oxygenase
- FGF, fibroblast growth factor
- HIF, hypoxia inducible factor
- IL, interleukin
- mAb, monoclonal antibody
- OA, osteoarthritis
- RA, rheumatoid arthritis
- SCID, severe combined immunodeficient
- SDF, stem cell derived factor
- TGFβ, transforming growth factor β
- TNFα, tumour necrosis factor α
- VCAM, vascular cell adhesion molecule
- VEGF, vascular endothelial growth factor
A ngiogenesis, the growth of new blood vessels, is important in a variety of fibroproliferative disorders, such as diabetic retinopathy, psoriasis, tumour growth, and rheumatoid arthritis (RA). We and others have reviewed this subject recently.1–15 Blood vessel growth probably contributes to the proliferation of the inflammatory synovial pannus as well as to the ingress of inflammatory leucocytes into the synovial tissue. The process of angiogenesis is a fine tuned balance between angiogenesis induction and inhibition. In this article, I will review some of the current advances in the angiogenesis field, specifically focusing on understanding angiogenesis in RA (table 1).
Some angiogenesis inducers relevant to RA
FIBROBLAST GROWTH FACTORS
Among the earliest identified angiogenic factors were the fibroblast growth factors. Folkman and colleagues took advantage of the observation that mast cells, which secrete heparin, were often found in proximity to blood vessels.16 Hence, the ability to bind heparin was used as the first method of isolating angiogenic factors. Recently Yamashita and coworkers found that the synovium of patients with RA and joints from rats with adjuvant induced arthritis (AIA) contained increased amounts of fibroblast growth factor-2 (FGF-2).17 Moreover, a Sendai virus containing the FGF-2 gene resulted in worse arthritis in rat AIA, but had no effect on normal joints. Hence, it seems that FGF-2 is important in worsening the progression of experimental arthritis rather than being important in the initiation stage of disease.
VASCULAR ENDOTHELIAL GROWTH FACTOR (VEGF)
An angiogenic mediator which has attracted much attention recently is VEGF, which is an endothelial selective growth factor.18 VEGF induces vascular permeability as well. In human RA, several groups have described VEGF in the joints and serum of patients.19–28 In the control of VEGF secretion in the RA joint, not only do cytokines like interleukin 1 (IL1) and tumour necrosis factor α (TNFα) induce fibroblast expression of VEGF but so also does engagement of the CD40 ligand.29,30 VEGF induces endothelial decay accelerating factor, which is cytoprotective against activated complement and may regulate endothelial proliferation and angiogenesis.31
VEGF binds to VEGF-R1/Flt-1 and VEGF-R2/Flk-1/KDR. Recently VEGF-C and VEGF-D have been described. In contrast with VEGF, VEGF-C is inducible by IL1/TNFα, but not hypoxia. VEGF-C is chemotactic for monocytes and mediates angiogenesis and lymphangiogenesis (the growth of lymphatic vessels in mouse skin). VEGF-C is the ligand for the FMS-like tyrosine kinase receptor Flt-4 (VEGF-R3). VEGF-C is strongly expressed in RA synovial lining, pericytes, and vascular smooth muscle.32 VEGF-D binds VEGF-R2 and VEGF-R3 on lymphatic endothelial cells and is angiogenic. VEGF-D expression is not abundant in RA synovial tissue.32 In the same study both VEGF-R2 and -R3 were expressed in RA synovium on blood vessels. Whereas VEGF-R3 has been considered to be relatively specific as a marker for lymphatic vessels in adult tissue, the significance of RA synovial endothelial VEGF-R3 expression is unclear. One may speculate that because synovial tissue contains fenestrated blood vessels, which play a part in nutrition of the avascular hyaline articular cartilage, VEGF-R3 may potentially be involved in the maintenance of fenestrations and formation of joint synovial fluid.
A number of drugs used to treat RA may modulate VEGF production. For instance, cyclosporin treatment of RA synovial fibroblasts exposed to transforming growth factor β (TGFβ) results in decreased production of VEGF.33 In patients with RA treated with anti-TNFα, vascular deactivation occurs so that serum levels of VEGF fall along with clinical improvement.34 Experimentally, modulation of a VEGF receptor through a VEGF-R1 Fc molecule suppressed RA synovial endothelial proliferation.35 The angiogenesis inhibitor TNP-470, a derivative of the fungal product fumigillin, suppressed angiogenesis and RA synovial tissue volume in a human RA synovial tissue-severe combined immunodeficient (SCID) mouse chimera, in which human RA synovium is allowed to implant and thrive in these.36 These studies suggest that drugs or potential future therapeutic agents may be useful in their ability to target VEGF.
The role of VEGF has been examined in several animal arthritis models also. Anti-VEGF reduces arthritis onset and severity as well as joint angiogenesis in mouse collagen induced arthritis (CIA).37,38 Preventative administration of TNP-470 in spontaneous models of RA in KRN×NOD mice results in attenuated arthritis and decreased serum VEGF.39 Other therapeutic approaches have been to target the VEGF receptor Flt-1. Hence, Miotla and coworkers and Luttun and coworkers have shown that anti-Flt-1 or soluble Flt-1 reduced synovial angiogenesis and arthritis.40,41 Administration of the fungal derivative TNP-470 in rat arthritis leads to attenuated arthritis and serum VEGF production.36
ROLE OF HYPOXIA INDUCIBLE FACTOR (HIF) IN ARTHRITIS
VEGF is inducible by hypoxia, which may occur in the inflamed joint.22,29 HIF-1, which comprises HIF-1α, and hydroxycarbon nuclear translocator (ARNT) controls many transcriptional responses to hypoxia by binding to hypoxia response elements of VEGF and other target genes.42 Hypoxia increases VEGF production by RA synovial fibroblasts, but this production is even more marked in the presence of inflammatory cytokines such as IL1 and TGFβ.29 These findings suggest that inflammatory cytokines augment the production of VEGF in the face of a hypoxic joint. Recently, Hollander and coworkers have identified HIF-1α in RA synovial tissue, predominantly in macrophages.43 Moreover, Cramer and colleagues showed that when the HIF-1α gene was deleted in mice, arthritis was attenuated in the KRN×NOD mouse arthritis model.44
HIF is thus becoming increasingly important in the pathogenesis of RA.
IL18 AS AN ANGIOGENIC MEDIATOR
Clearly cytokines such as TNFα and VEGF are important angiogenic mediators in RA. We have recently defined a new cytokine, IL18, in the induction of RA synovial angiogenesis.45 IL18 is a potent inducer of endothelial chemotaxis in vitro and angiogenesis in vivo in the Matrigel plug assay and sponge angiogenesis assay in rodents. In the Matrigel plug assay (fig 1), the matrix protein Matrigel containing an angiogenic mediator is implanted in mice. After 7–10 days the plugs are harvested and blood vessel growth quantified by histology or assay of plug homogenate for haemoglobin, which is proportional to the number of blood vessels. Immunodepletion of IL18 from RA synovial fluids resulted in reduced endothelial migration in vitro, suggesting that IL18 is important in RA mediated angiogenesis. IL18 appears to act on endothelium through an αvβ3 mediated mechanism (see below).

Schematic diagram of the Matrigel angiogenesis in vivo assay.
CHEMOKINES AS ANGIOGENIC MEDIATORS
Among other important mediators of angiogenesis are chemokines. Most chemokines are low molecular weight (8–10 kDa) proteins, which are predominantly known for their ability to recruit leucocytes. Chemokines are divided into the C-X-C, CC, and C-X3-C families based on the presence or absence of an amino acid, X, between a pair of cysteine residues near the amino terminus of the protein. In collaboration with Dr Robert Strieter, Peter Polverini, and Steve Kunkel, our group found that monocyte/macrophage derived IL8, a prototype of the C-X-C chemokine subfamily, was angiogenic.46 This factor appeared important in that synovial tissue macrophage-derived chemotactic activity for endothelial cells in vitro and angiogenesis in vivo was significantly decreased if IL8 was immunodepleted.
In general, chemokines like IL8, of the C-X-C class containing the amino acid E-L-R motif are angiogenic, while those lacking this motif are angiostatic.47 Exceptions to this generalisation exist in that the C-X-C chemokine stem cell derived factor-1 (SDF-1), which lacks the E-L-R motif, is angiogenic.48 Hitchon and colleagues have recently shown that hypoxia up regulates both VEGF and SDF-1 expression in RA synovial fibroblasts.49 Clearly chemokines, such as SDF-1, also recruit leucocytes to the inflamed RA synovium50,51
We have shown that fractalkine is the first chemokine described of the CX3C class to mediate angiogenesis.52 Fractalkine is so called for its fractal geometry and is the sole member of the CX3C class of chemokines.53 It contains a chemokine motif on top of a mucin-like stick, the so-called chemokine on a stick.53 Fractalkine is a unique chemokine in that it can also act as an adhesion molecule when cell bound. Fractalkine induces endothelial tube formation on the matrix Matrigel in vitro. Similarly, fractalkine induces angiogenesis in Matrigel plugs implanted in mice in vivo. When fractalkine is immunodepleted from RA synovial fluids, the ability of these synovial fluids to chemoattract endothelial cells, a facet of the angiogenic response in vitro, is decreased. Hence, chemokines are probable contributors to RA angiogenic activity.
MECHANISM OF ACTION OF SOME ANGIOGENIC CYTOKINES: THROUGH ADHESION MOLECULES LIKE INTEGRINS
The invasion, migration, and proliferation of endothelial cells is regulated, at least in part, by the integrin family of cell adhesion molecules. αvβ3 is minimally, if at all, expressed on resting or normal blood vessels but is highly expressed in RA synovial blood vessels.54 Some angiogenic factors like basic fibroblast growth factor (bFGF), TNFα, and IL18 may act through αvβ3 integrins.55 VEGF or TGFα appear to act through an αvβ5 integrin mechanism using protein kinase C. This may turn out not to be a main mode of action of VEGFs in RA because αvβ5 integrin has been reported to be expressed in normal and osteoarthritic (OA) synovial tissue, but not RA synovial tissue.56 None the less, the mechanisms by which some of these cytokines act to promote angiogenesis are rapidly becoming identified.
αvβ3 has been targeted using animal arthritis models. Storgard and colleagues showed that anti-αvβ3 integrin decreased synovial angiogenesis in a rabbit arthritis model.57 An oral non-peptide αvβ3 antagonist ameliorated arthritis in rats.58 Finally, a proaptotic αvβ3 antagonist, composed of an RGD peptide linked to a heptapeptide dimer, is therapeutic in mouse CIA and selectively homes to mouse arthritic synovium compared with normal synovium and with control organ endothelium.59 Clearly, this integrin is a promising target for angiogenesis inhibition.
SOLUBLE ADHESION MOLECULES AS ANGIOGENIC MEDIATORS
Endothelial cells express soluble adhesion molecules, particularly upon cytokine stimulation. Cellular adhesion molecules are shed from the cell surface and secreted. The function of these soluble adhesion molecules is unclear. Neidhardt, Gay, and colleagues have found that the new endothelial antigen CD146 (Muc18), which is potentially involved in endothelial-leucocyte adhesion, is increased in the soluble form in RA compared with OA synovial fluids.60
We have examined other soluble adhesion molecules. A prevailing paradigm was that these molecules might serve an anti-inflammatory role by binding leucocytes, thus preventing them from adhering to endothelium and entering inflamed tissues. We proposed the converse paradigm that these molecules may be proinflammatory in some cases. Our laboratory found that the cellular adhesion molecules soluble E-selectin and soluble vascular cell adhesion molecule-1 (VCAM-1) are angiogenic.59,61,62 It is likely that activated cells in the synovial milieu, such as endothelial cells, bear E-selectin and VCAM-1, which they then shed into the synovial fluid. These soluble adhesion molecules then interact with vascular endothelial cells through sialyl Lex in the case of soluble E-selectin, and VLA-4, in the case of soluble VCAM-1 to mediate angiogenesis.59,61,62 We have recently shown that soluble E-selectin acts on endothelial cells through an Src and phosphoinositol-3 kinase mediated pathway.62a Inhibition of these pathways by chemical inhibitors, as well as antisense oligodeoxynucleotides directed against these signalling molecules, inhibited angiogenesis in both in vitro and in vivo assays. Interestingly, patients with RA treated with monoclonal anti-TNFα have an element of endothelial deactivation, in which treatment reduces levels of soluble E-selectin.63 Hence, these findings may be important clinically in RA.
GLYCOCONJUGATES AS ANGIOGENIC MEDIATORS
We have described another new related angiogenic mediator in our laboratory. This antigen, termed 4A11, is an endothelial selective, cytokine inducible, endothelial angiogenic antigen.64 We first raised monoclonal antibody (mAb) 4A11 by immunising mice with isolated adherent cells from human RA synovial tissue. The mAb we produced recognised endothelium in the synovium, thymus, skin, and lymph node selectively, perhaps suggesting a role in cell homing to these regions. Moreover, the mAb was endothelial selective, recognising endothelium and keratinocytes only. This antigen is up regulated in RA compared with normal synovial tissue and is rapidly cytokine inducible in vitro, being stored in cytoplasmic vesicles and up regulated on the cell surface within 5–20 minutes of contact with cytokines. We have obtained a partial structure of the antigen recognised by mAb 4A11. The mAb detects Lewisy-6 and H-5-2 antigens (Ley/H). These structures are mainly recognised for their function as blood group antigens. Interestingly, these antigens are structurally related to the E-selectin ligand sialyl Lewisx. Because of the angiogenic properties of soluble E-selectin, we suggested these endothelial antigens were released by activated endothelium and induced angiogenesis (fig 2). Glucose analogues of these molecules or the glycolipids themselves induced a potent endothelial chemotactic response. Moreover, the glucose analogues are angiogenic in vivo in the corneal bioassays. mAb 4A11 abrogated the angiogenic responses. We reasoned that if these molecules mediated angiogenesis, they might be detected in clinical samples from patients with RA. We found that soluble 4A11 antigen is increased in RA compared with OA serum and synovial fluid. These results describe a new endothelium-selective antigen that functions as an angiogenic mediator. As with the E-selectin and VCAM-1, it is likely that endothelial cells exposed to cytokines bear the 4A11 antigen, which is shed in the inflamed joint and mediates angiogenesis.

Suggested mechanism of the soluble 4A11 antigen (Ley/H). Endothelial cells are activated by inflammatory mediators to produce the 4A11 antigen, which is then secreted to mediate angiogenesis in adjacent endothelial cells. This is the same paradigm for soluble molecules such as CD146, soluble E-selectin, or soluble VCAM-1.
Glycoconjugates (glycoproteins/glycolipids) have been known for some time to constitute the chemical basis for several blood group systems in man and to act as adhesion molecules for microbial ligands.65 The importance of glycoconjugates in the induction of autoimmunity was recently emphasised by the finding that Helicobacter pylori, the micro-organism involved in gastritis, ulcers, adenocarcinoma, and lymphoma of the stomach, expresses Ley/Lex/H, which is also found in gastric mucin.66,67 Mice bearing hybridomas making H pylori induced anti-Le antibodies developed gastritis, pointing to a mechanism by which H pylori participates in molecular mimicry. Thus, antibodies directed against H pylori Ley result in gastritis through an autoimmune reaction directed against gastric mucin Ley. In diseases such as RA the inciting agent is unknown. Thus, possibly, Ley/H may also trigger a molecular mimicry immune reaction in inflammatory angiogenic sites such as RA.
One hypothesis suggests that while endothelium is quiescent for weeks or longer, endothelial cells must also require a mechanism of storage of preformed regulators of angiogenesis which can induce new capillary growth within hours in response to angiogenic stimuli, such as those found in a wound or an inflamed synovial tissue.68 Despite this hypothesis, with the possible exception of bFGF, examples have not been described for angiogenesis inducers. The rapid cell surface expression of Ley/H may fit this paradigm. However, an alternative scenario for the regulation of angiogenesis by inducers and inhibitors may be that structural mimicry has a role. Hence, for instance, a search based on crystal structure showed that the angiogenic inhibitor endostatin was most homologous to the angiogenic mediator E-selectin.69 It is likely that in an RA joint Ley/H may be stored for expedient use during times of active inflammation and subsequent angiogenesis.
THE ANGIOPOIETIN-TIE SYSTEM
Unlike VEGF, angiopoietin-1 (Ang-1) is not a mitogen for endothelium, nor does it induce endothelial tube formation in vitro. Instead, its pattern of expression in the vicinity of forming vessels suggests that it has a role in regulating the assembly of non-endothelial vessel wall components like vascular smooth muscle.70,71 This finding is strengthened by the observation of Suri and coworkers, who showed that mice deficient in Ang-1 exhibit abnormal vasculature where the principal defect is failure to recruit vascular smooth muscle cells. For instance, in the heart, this defect is manifest as poorly developed endocardium.72 In RA, we and Gravallese and coworkers have shown that Ang-1 is expressed in human RA synovium in lining cells, macrophages, fibroblasts, and endothelium.73,74 Expression of Ang-1 is higher in RA than in OA synovial tissue.73 TNFα can serve as a stimulus to induce Ang-1 expression in RA synovial fibroblasts.74
Ang-2 counters the effects of Ang-1 by disrupting blood vessel formation in the developing embryo by antagonising Ang-1 induced phosphorylation of the Tie-2 receptor, to which Ang-1 and Ang-2 bind. Ang-2 is also up regulated in RA compared with OA synovial tissue.73
One of the most exciting findings described by Vikkula and coworkers, was the discovery that venous malformations in two human families could be mapped to the Tie-2 receptor on endothelial cells, where a missense mutation results in an arginine to tryptophan substitution.75 Patients carrying this mutation develop vein-like structures that are deficient in the non-endothelial cells of the vessel wall; mainly they lack vascular smooth muscle cells, resulting in thin walled vessels Therefore, the Tie-2/Ang system seems to govern maturation and stabilisation of blood vessels.
Whereas Tie-2 binds both Ang-1 and Ang-2, Tie-1 is an orphan receptor. In our study, Tie-1 expression was found on about 75% of synovial endothelial cells in RA compared with only 3% of endothelial cells in OA synovium.73 Tie-1 expression was not endothelial-specific, being expressed by a variety of other synovial cell types.73,76 Like Tie-1, Tie-2 is also expressed on a variety of cells in the synovium and up regulated in RA compared with OA.73,76 Hence, the delicate balance between members of the Ang and Tie families may contribute to vascular formation in RA (table 2).
Some angiogenesis inhibitors relevant to RA
PARADIGM OF INHIBITORS RESIDING WITHIN LARGER PROTEINS
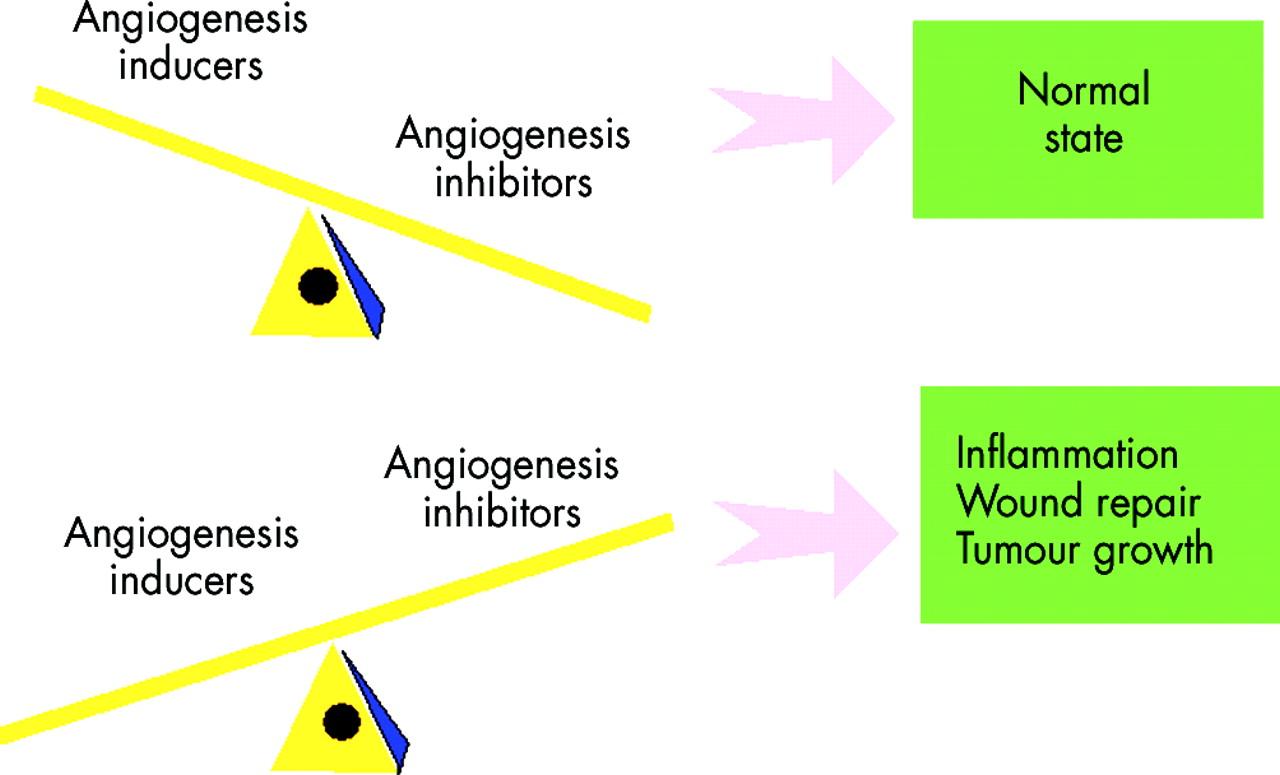
Counterbalancing the angiogenesis inducers are the angiogenesis inhibitors (fig 3). One paradigm regarding angiogenesis inhibitors, mentioned above, is that angiostatic activity often resides in portions of larger common proteins, which may or may not themselves be angiostatic.68 Examples of this include many molecules thought to have a role in RA pathogenesis, like thrombospondin, fibronectin, and propeptides of type II collagen, platelet factor IV, and fragments of epidermal growth factor. Other mediators fitting this paradigm are the 29 kDa fragment of fibronectin, the 16 kDa fragment of prolactin, and angiostatin (a fragment of plasminogen), among others.77–80

{kind=link}
{kind=link}
{kind=link}
Balance of angiogenesis inducers and angiogenesis inhibitors in pathological disease states.
THROMBOSPONDIN AS AN ANGIOGENESIS INHIBITOR
The idea that endothelium is quiescent for long periods of time and yet can be induced to sprout new capillaries in a matter of hours in response to an angiogenic stimulus, suggested that angiogenesis regulators might be stored for expedient use. The first indication of this paradigm was described by Bouck and coworkers, who found that a non-tumorigenic hamster cell line became tumorigenic with a mutation that inactivated a tumour suppressor gene.81 The inhibitory activity was found to be a fragment of the adhesive glycoprotein thrombospondin-1 whose expression was linked to the presence of a tumour suppressor gene. Thrombospondin appears to act by inducing endothelial cell apoptosis.82 Recently, metallospondins have been found using molecular techniques, and are even more potent angiogenic inhibitors than thrombospondin.83 We were unable to show that they inhibited arthritis or angiogenesis in a rat model of AIA.83,84
ANGIOSTATIN AS AN ANGIOGENESIS INHIBITOR
Another inhibitor of angiogenesis termed angiostatin has been identified in some very elegant studies by Dr Judah Folkman’s group.78,85–89 This factor is a potent inhibitor of tumour growth. Angiostatin is a fragment of the clotting factor plasminogen. Plasminogen itself is not angiostatic. This factor acts by depleting energy required for blood vessel growth by binding ATP-synthase and induces endothelial cell apoptosis by activating focal adhesion kinase.88–90 Recently, angiostatin delivered through a retroviral vector, has been shown to reduce angiogenesis and CIA in mice.91 This study lends hope to the notion that endogenous angiostatic factors may be of therapeutic benefit in RA.
ENDOSTATIN AS AN ANGIOGENESIS INHIBITOR
Endostatin, an angiogenesis inhibitor produced by mouse haemangioendothelioma cells, is a fragment of collagen type XVIII.92 Thus, the fact that abundant components of the circulatory system such as fibronectin and plasminogen can be converted to potent angiostatic factors suggests a new form of regulation by proteases, such as serine proteases, to specifically release these molecules from their parent molecules. There has been recent interest in endostatin therapy in models of arthritis. Nagashima and coworkers showed that endostatin levels are not increased in RA compared with OA synovial fluids.93 When these investigators administered endostatin in an RA synovial tissue-SCID chimera model, inflammation and angiogenesis were reduced in the synovium.36 Yin and coworkers used lentiviral gene therapy to deliver endostatin to TNFα transgenic mice, which spontaneously develop arthritis.94 Endostatin decreased mouse joint mRNA levels of a number of angiogenic factors, including VEGF and bFGF. Most cytokines that were decreased seemed to use the Jun kinase (JNK) signalling pathway.
ENDOGENOUS INHIBITOR OF ANGIOGENESIS DERIVED FROM CARTILAGE
Interestingly, cartilage is avascular as well as relatively tumour resistant. So, it is reasonable that among the earliest described inhibitors of neovascularisation were cartilage derived inhibitors. Inhibitors of angiogenesis have been purified from various types of cartilage.95–99 Shark cartilage contains a potent inhibitor of angiogenesis, accounting for its popularity in some circles as an unorthodox treatment for cancer.100 A cartilage derived inhibitor from bovine scapula has been described which is also an inhibitor of collagenase. Recently, a human cartilage derived angiogenesis inhibitor has been shown to be troponin I, a protein responsible for regulation of muscle contractions.95,96,101 Like angiostatin, troponin I acts by binding ATPase, thus depleting energy needed for blood vessel growth. Regardless of the source, it is intriguing that cartilage, which lies in juxtaposition to the inflamed RA synovium, may hold the key to an understanding of how one might inhibit the angiogenic process in the joint.
CYTOKINES AS ANGIOSTATIC AGENTS
A number of cytokines are potential angiostatic agents. For instance, we and others have shown that IL4 is of therapeutic benefit in animal models of RA and in human RA synovial tissue explant cultures.102–104 In our hands, a portion of the mechanism of this cytokine in rat AIA is through reduction of synovial angiogenesis.103,105
Recently an inhibitor of IL1, the IL1 receptor antagonist has been shown to block corneal angiogenesis in the rat.106 Moreover, this cytokine reduced the number of blood vessels and decreased arthritis in joints of rats with AIA.
THALIDOMIDE AS AN ANGIOSTATIC AGENT
Thalidomide was once marketed as a sleeping pill until it was shown to induce defective limb development in the offspring of pregnant females who ingested the drug. This defect was probably due to its ability to inhibit angiogenesis, perhaps by binding TNFα, which is a key mediator in RA. In animal studies, it appears that thalidomide suppresses the severity of rat CIA, but not through TNFα or VEGF down regulation.107,108 Thalidomide decreased inflammation and numbers of blood vessels in a human RA synovial tissue-SCID chimera.36 Thalidomide has been used in two small trials in patients with refractory RA, either alone or in combination with pentoxyfilline.109,110 Although this treatment was somewhat effective, the side effect profile was considerable, with adverse reactions including drowsiness, xerostomia, and constipation.
FUMIGILLIN AND ITS DERIVATIVES AS ANGIOSTATIC AGENTS
Fumigillin is an angiostatic compound discovered as a fungal contaminant on an endothelial culture dish, which inhibited endothelial growth.111–114 Derivatives of this compound termed AGM-1470 or TNP-470 inhibit both angiogenesis and arthritis in rat AIA and CIA models, as well as in the KRN×NOD model in mice.39,114,115 Interestingly, angiogenesis may also have a role in bone formation. TNP-470 inhibits ectopic bone formation induced by bone morphogenetic protein in mice.116 Fumigillin may be a potential therapeutic agent, inhibiting arthritis and angiogenesis in RA.
SULFASALAZINE AS AN ANGIOSTATIC AGENT
Sulfasalazine is commonly used in the treatment of a variety of diseases, including RA. The active metabolite of sulfasalazine may be sulfapyridine. In one study, while sulfapyridine inhibited endothelial proliferation, sulfasalazine and 5-aminosalicylic acid metabolites did not.117 We have shown that sulfasalazine and sulfapyridine reduce endothelial proliferation as well as chemotaxis.118 Sulfapyridine also inhibits phorbol myristate acetate-induced endothelial tube formation in vitro. We have also shown that sulfapyridine may mediate this effect by reducing endothelial IL8 production.
OTHER ANGIOSTATIC AGENTS
Some of the endogenous and exogenous inhibitors of angiogenesis which have been identified to date include angiostatic corticosteroids, minocycline, choloroquine, methotrexate, penicillamine, TNFα inhibitors, thiol-containing compounds such as gold compounds, taxol, 2-methoxyoestradiol, and cyclo-oxygenase-2 (COX-2) inhibitors.
Several angiogenesis modulating agents have recently been tried in rodent models of arthritis. These include taxol and 2-methoxyoestradiol. Taxol, a chemotherapeutic agent, inhibited CIA in rats and inhibited synovial angiogenesis.119 2-Methoxyoestradiol treatment of CIA arthritis in mice reduced the arthritis. In vitro, this compound inhibited proliferation of endothelial cells.120
Prostaglandins, such as prostaglandin E2, are potent mediators of angiogenesis.121 COX-2 inhibitors are used for the treatment of RA. Not surprisingly, they have been shown to inhibit angiogenesis mainly in tumour models.27,122–125 Possibly, these modulators are also effective in reducing RA angiogenesis.
CAN ANGIOGENIC MARKERS HELP GUIDE TREATMENT IN RA?
Some angiogenic markers may be useful. For instance, soluble CD146 is up regulated in synovial fluids from patients with RA compared with traumatic injury.60 Patients with early RA had the highest values of this marker in their synovial fluid. Soluble CD146 correlated significantly with the degree of morning stiffness, the number of tender joints, and the number of swollen joints.
Fraser and coworkers recently used needle arthroscopy in 34 patients, of whom 12 had early RA. They showed that synovial fluid matrix metalloproteinase 9 levels correlated with blood vessel morphology and synovial fluid VEGF levels, particularly in early RA.126 These studies indicate that angiogenic markers have the potential to help guide us in RA management.
CAN ANGIOGENIC MARKERS PREDICT RA DISEASE OUTCOME?
In a study of 44 patients, serum VEGF at presentation in patients with early RA correlated significantly with the development of radiographic damage after one year.127 This study is perhaps the most compelling one to depict a role for angiogenesis modulation guiding treatment for RA.
WHAT CAN WE EXPECT IN ANGIOGENESIS MODULATION IN THE FUTURE?
A new notion has developed over the recent few years that preferential endothelial precursors may exist within the population of blood stem cells. For instance, some CD34+ cells carrying receptors for VEGF may, under some circumstances, develop into endothelial cells.128–130 These cells may be important in the induction and perpetuation of angiogenesis, as well as endothelial differentiation.128–130 In addition, they may also be used for the induction of neovascularisation in future therapeutic trials in certain vascular disorders.131–133 VEGF and bFGF have been used in clinical trials to induce angiogenesis, by stimulating endothelial morphogenesis from stem cells in ischaemic heart disease,131 as well as obliterative arteriosclerosis.132,133 The use of this technology to modulate angiogenesis has not yet been used in RA or its animal models, but may hold promise in arthritis as well.
Alternatively, one may envision a future where additional angiostatic agents are found that confer minimal toxicity when used as therapeutic agents. Other strategies may be the administration of angiostatic cytokines through genetic or viral approaches. Some targets may include receptors for angiogenic factors as the hierarchy of angiogenic and angiostatic factors in RA becomes established. Regardless of the approach, data in recent years favour the modulation of the angiogenic balance as a viable therapeutic option for RA.
Acknowledgments
Support for this work was provided by NIH grants AI40987 and HL58695, the Veteran’s Administration Research Service, and the Gallagher Professorship for Arthritis Research. Apologies to those whose work was not cited owing to space limitations.
REFERENCES
- 1.↵
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.
- 21.
- 22.↵
- 23.
- 24.
- 25.
- 26.
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 62a.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.
- 87.
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.
- 98.
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.
- 113.
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.
- 124.
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.
- 130.↵
- 131.↵
- 132.↵
- 133.↵