Article Text

Abstract
Background: Mutilating sensory neuropathy with spastic paraplegia is a very rare disease with both autosomal dominant and recessive modes of inheritance. We previously mapped the locus of the autosomal recessive form to a 25 cM interval between markers D5S2048 and D5S648 on chromosome 5p. In this candidate interval, the Cct5 gene encoding the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (CCT) was the most obvious candidate gene since mutation in the Cct4 gene encoding the CCT delta subunit has been reported to be associated with autosomal recessive mutilating sensory neuropathy in mutilated foot (mf) rat mutant.
Methods: A consanguineous Moroccan family with four patients displaying mutilating sensory neuropathy associated with spastic paraplegia was investigated. To identify the disease causing gene, the 11 coding exons of the Cct5 gene were screened for mutations by direct sequencing in all family members including the four patients, parents, and six at risk relatives.
Results: Sequence analysis of the Cct5 gene revealed a missense A492G mutation in exon 4 that results in the substitution of a highly conserved histidine for arginine amino acid 147. Interestingly, R147 was absent in 384 control matched chromosomes tested.
Conclusion: This is the first disease causing mutation that has been identified in the human CCT subunit genes; the mf rat mutant could serve as an animal model for studying these chaperonopathies.
- CCTε
- chaperonin containing TCP-1 epsilon subunit
- missense mutation
- mutilating sensory neuropathy
- spastic paraplegia
Statistics from Altmetric.com
- CCTε
- chaperonin containing TCP-1 epsilon subunit
- missense mutation
- mutilating sensory neuropathy
- spastic paraplegia
Mutilating hereditary sensory neuropathy with spastic paraplegia is a very rare disease with both autosomal dominant1–3 and autosomal recessive4–7 modes of inheritance. The autosomal recessive form is characterised by progressive distal sensory neuropathy, often complicated by severe infections, osteomyelitis, and amputations, and by a relatively mild paraplegia related to bilateral upper motor neuron involvement. Some clinical characteristics differentiate the patients described by Cavanagh et al4 from those described by Thomas et al5 in terms of disease onset, atrophy of the spinal cord, and defects in upper limbs, suggesting the existence of genetic heterogeneity in this group of disorders.
We recently mapped the disease locus in a consanguineous Moroccan family (family IDR) with mutilating sensory neuropathy associated with spastic paraplegia to a 25 cM genetic interval between markers D5S2054 and D5S648.8 In this genetic interval, where more than 90 genes and pseudogenes have been mapped, the most obvious functional candidate gene seems to be the Cct5 gene encoding the epsilon subunit of the cytosolic chaperonin-containing t-complex peptide-1 (CCT complex) since chaperones have been associated with neurodegenerative disease.9 Also, mutation in the Sprague-Dawley rat Cct4 gene, which encodes the delta subunit of the CCT complex, has been shown to be associated with mutilating sensory neuropathy,10 whereas mutation in the CCT gamma subunit has been reported to cause degeneration of retinal neuroepithelial cells in zebrafish.11
In the present study, we report a missense mutation in exon 4 of the Cct5 gene as the cause of mutilating sensory neuropathy with spastic paraplegia.
METHODS
In the Department of Neurology and Neurogenetics, Hôpital des Spécialités (Rabat, Morocco), we identified a Moroccan consanguineous family (IDR) with four affected males displaying a mutilating sensory neuropathy with spastic paraplegia. The disease onset was in early infancy with spastic paraplegia and progressive sensory loss leading to mutilating acropathy involving both upper and lower limbs. Magnetic resonance imaging revealed severe atrophy of the spinal cord predominantly in the posterior tract.8
Blood samples were obtained with the informed consent of all IDR family individuals in accordance with study protocols approved by the university ethics committees. Genomic DNA was extracted using standard procedures. The Cct5 gene (ENSG00000150753) consists of 11 exons, and exon specific intronic primers were designed covering the splice sites at both ends of the exons (table 1). Both strands were sequenced with the BigDye dRhodamine Terminator Reaction Kit (version 1.1) according to the manufacturer’s instructions using an automated ABI 310 DNA sequencer. The collected chromatogram data were analysed with SeqScape software version 2.0 (all from Applied Biosystems, Foster City, CA).
PCR primers for sequence analysis of the Cct5 gene
RESULTS AND DISCUSSION
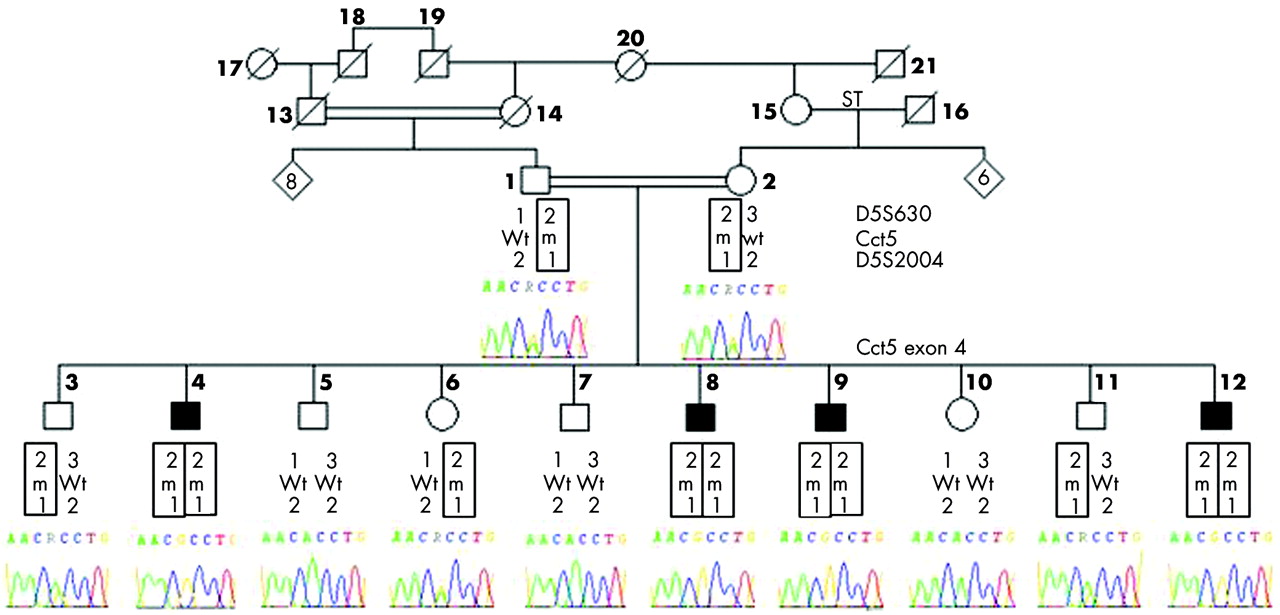
We PCR amplified and directly sequenced the entire coding region of the Cct5 gene, including exon-intron boundaries, in the proband of the IDR family. We identified a homozygous mutation in exon 4 which consists of an A492G variation that generates a missense mutation H147R. Sequencing of Cct5 exon 4 in the other members of the IDR family confirmed that the variant allele segregated with the disease (fig 1) and was absent from 384 ethnically matched control chromosomes.

Segregation of the mutation A492G in the pedigree of the IDR family displaying autosomal recessive mutilating sensory neuropathy with spastic paraplegia. Black symbols identify affected individuals; circles represent females, and squares males. Genotypes for D5S630, Cct5 exon 4, and D5S2004 are indicated below individuals: Wt, wild type; m, mutated allele. Electrophoregrams of the Cct5 exon 4 sequence from positions 489 to 495 are shown for all individuals tested.
The Cct5 gene spans 14.79 kb with 11 exons that encode the epsilon subunit of the CCT complex, a 541 amino acid polypeptide. Comparison of the human CCTε protein sequence with those in the peptide sequence database indicates that the H147, the substituted amino acid in the studied family, is highly conserved across diverse groups of species (fig 2).

{kind=link}
{kind=link}
Clustal W alignment of the Cct5 orthologs of the region surrounding the H147R missense mutation.
Molecular chaperones play an important role in the folding of many proteins and the CCT complex is a member of the two major chaperone systems that promote the folding and assembly of a wider range of cytosolic substrates. CCT particles consist of two stacked rings of eight or nine subunits each, forming a large cylindrical complex with a central cavity in which the folding of proteins, particularly actin and tubulin, occurs in an ATP dependent process.12 All CCT subunits, except the testis specific subunit ζ-2, were reported to be expressed in many tissues, and all are required for CCT to function.13
The peptide sequences of all CCT subunits contain 30% amino acid identity and highly conserved motifs for ATP binding, but they diverge in the putative polypeptide binding domains.14 It has been proposed that each subunit recognises specific proteins and the complex ATPase function could be collectively modulated by all CCT subunits.15
The identified H147R missense mutation in the studied IDR family is located in the equatorial domain 1, predicted by analogy to the prokaryote GroEL structure,16 a homolog of the human group I chaperonin HSP60 that has been implicated in hereditary spastic paraplegia.17 It is unclear how the H147R mutation causes mutilating sensory neuropathy with spastic paraplegia. However, since it has been reported that the equatorial domain could be involved in ATP binding and the apical domain in substrate binding,16 the mutation may affect the binding and hydrolysis of ATP, affecting the epsilon subunit ability to bind and fold its specific substrates, mainly actin and tubulin, the two major proteins folded by the CCT complex. Misfolding of these cytoskeleton proteins by the defect in the ATP binding and folding activity of the CCT epsilon subunit may cause the defect in their slow axonal transport and their assembly into microfilaments as reported by Bourke et al,18 leading to apoptosis. This mechanism is in accordance with the findings that in all patients of the IDR family, the sensory neuropathy was distal with ulcerations starting in toes and fingers, and neuronal apoptosis in the spinal cord.8
It has been shown previously that actin and tubulin bind to CCT in geometric arrangements involving interactions of their large C-terminal domains with CCTε/β and their small N-terminal domains with CCTδ/θ subunits.19,20 This finding can be corroborated by the fact that mutation in the CCTδ subunit in mf rat mutant causes a similar disease phenotype as in our IDR family, with mutilating sensory neuropathy10 and excessive neuronal apoptosis in the dorsal root ganglia.21 This mf rat mutant could serve as an excellent animal model for studying the physiopathological mechanism of the CCT complex.
In conclusion, we present the first report showing a mutation in the epsilon subunit of the human CCT complex as a cause of autosomal recessive mutilating sensory neuropathy with spastic paraplegia. Reports of other mutations in the different CCT subunits will offer the opportunity to increase our understanding of this CCT chaperonin assisted folding in the cell, which might lead to effective treatment for these chaperonopathies.
Acknowledgments
We thank all family members for their cooperation throughout this study.
REFERENCES
Footnotes
-
Published Online First 19 January 2006
-
All authors of this paper are members of the PCNG network “Pôle de Compétences en Neurogénétique” of the Faculté de Médecine et de Pharmacie, of University Mohamed V Souissi (Rabat, Morocco)
-
This research was supported by the Presidency of the University Mohamed V Souissi (Rabat, Morocco) and the Association Marocaine de Neurogénétique (Morocco)
-
Competing interests: none declared