

Abstract
Osteoarthritis (OA) is a complex disease that affects the whole joint, with multiple biological and environmental factors contributing to its development. The heritable component for primary OA accounts for ∼50% of susceptibility. So far, candidate gene studies and genome-wide association scans have established 18 OA-associated loci. These findings account for 11% of the heritability, explaining a rather small fraction of the genetic component. To further unravel the genetic architecture of OA, the field needs to facilitate more precise phenotypic definitions, high genome coverage, and large sample metaanalyses, expecting the identification of rare and low frequency variants with potentially higher penetrance, and more accurate methods for calculating phenotype-genotype correlation. Expression analysis, epigenetics, and investigation of interactions can also help clarify the implicated transcriptional regulatory pathways and provide insights into further novel pathogenic OA mechanisms leading to diagnostic biomarker identification and new, more focused therapeutic disease approaches.
Osteoarthritis (OA) is a musculoskeletal disorder that causes degradation of synovial joints. It is the most common joint disease, and most frequently affects the hip, knee, spine, and the small joints of the hand and the foot. OA causes loss of articular cartilage, formation of osteophytes, changes in subchondral bone, and synovitis1. It is a complex disease that affects the whole joint, with multiple biochemical, genetic, biological, morphological, biomechanical, and environmental factors contributing to disease occurrence1,2. The heritable contribution to primary OA susceptibility has been established by twin and sibling studies to be about 50% (range 40–65%), and is transmitted in a non-Mendelian manner3. OA typically has its onset in mid-to-later life, indicating that age is a susceptibility risk factor. OA is more common in women, particularly following menopause. Sex differences may be partly explained by the suggested involvement of estrogen in cartilage metabolism4. OA is also thought to be associated with body mass index, joint injury, and lifestyle1. OA has a large effect on society. Psychological and physical disability lead to a substantial economic burden that is expected to grow with the increasing prevalence of OA5.
The prevalence of hip and knee OA varies by ethnicity. Asian populations show a higher prevalence for knee OA and a lower prevalence for hip OA in the Chinese and Japanese compared with populations of European descent6. These differences in OA prevalence may also indicate genetic heterogeneity. The interplay between genetic and environmental factors in OA is not fully understood and only a small fraction of the genetic component has been explained. The purpose of our perspective article is to review the current genetic epidemiological landscape of OA and to explore future opportunities to further unravel its genetic architecture.
CURRENT FINDINGS IN OA GENETICS
Genetic architecture of OA
As with other common complex disorders, the genetic architecture of OA is underpinned by variants covering a wide range of frequencies and effect sizes7. Most established OA-associated variants are represented by common single-nucleotide polymorphism (SNP) with minor allele frequency (MAF) > 5% that have moderate to small effect sizes (OR ∼1.1–1.3). The small number of OA loci established to date account for only a small fraction of disease heritability (10.7%; Figure 1)3. It is also possible to estimate the proportion of the genetic component explained by all variants using genome-wide association study summary statistics, with approaches such as Restricted Maximum Likelihood and Phenotype correlation genotype correlation regression8,9. An application in case-control studies suggests that common variants could explain at least half the heritability for common diseases. Low frequency and rare variants (MAF 1–5% and MAF < 1%, respectively), epigenetic changes, structural variants, and gene-environment interactions may also contribute to the missing heritability of OA10.
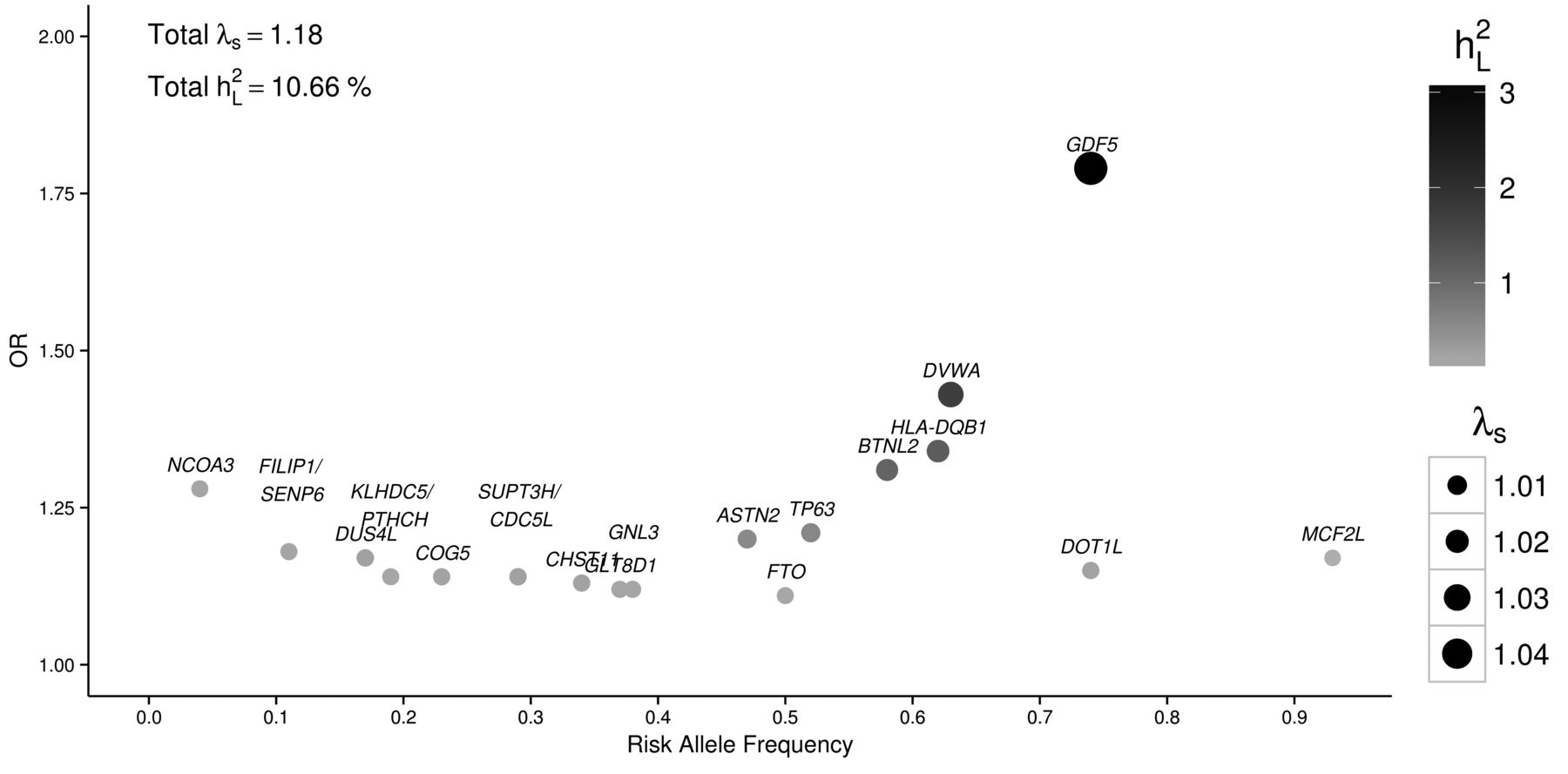
Effect size and RAF of established hip and knee OA-associated variants. Most SNP are common (RAF 0.04 to 0.93) with moderate to small effect sizes (OR 1.11 to 1.79) as reported in the respective studies. The sibling relative risk ratio (λs) for each SNP is shown by the different size of each circle. The darker color of the circles shows the percentage of variance explained on the liability scale (h2L; calculated assuming 10% prevalence of OA). Totals of λs and h2L are shown on the left top of the plot. RAF: risk allele frequency; OA: osteoarthritis; SNP: single-nucleotide polymorphism.
Established OA loci
Several linkage scans have been conducted for OA, but most have failed to establish replicating allelic loci. Numerous candidate gene studies based on the (to date) limited known biological etiology of OA have been conducted and have also been irreproducible3. Multiple factors, including lack of study power, account for this lack of replication. Exceptionally, GDF5 that has been associated repeatedly with OA was originally discovered in gene-centric studies. GDF5 affects chondrogenesis and joint element formation during skeletal development, which are consistent with the protein expression in the cartilage and joint interzone11. A candidate gene study first reported that rs143383 (in the 5′ untranslated region) was associated with hip OA in 2 independent Japanese populations (OR 1.79, p = 1.8 × 10−13)12. The SNP is associated with reduction in GDF5 transcription in chondrogenic cells, which results in reduced levels of protein expression. This locus was also later found to be associated with OA in European populations13. Genetic variation in GDF5 was also found to be associated with height14. It is also suggested that lower levels of GDF5 in mice include mechanisms of altered loading and changes in subchondral bone, exhibiting developmental failure of the condyles and the articular ligaments15 (Table 1).
Polymorphisms associated with hip and knee OA.
Genome-wide association studies (GWAS) examine hundreds of thousands of SNP across the genome and seek to establish associations with the risk of complex diseases and traits. The insights afforded by these studies can help improve our understanding of the key pathways involved in disease pathogenesis, and may lead to novel approaches to disease diagnosis, treatment, prevention, prognostic markers, and personalized (precision) therapy. In the field of complex disease, thousands of SNP have been robustly associated with disease risk at genome-wide statistical significance (p ≤ 5 × 10−8)16. Several small- and medium-scaled GWAS have been carried out for OA.
A GWAS identified a missense variant, rs7639618, in DVWA that is associated with knee OA, and replicated at a genome-wide significant level (p = 7.3 × 10−11) in a Japanese and combined Japanese and Han Chinese cohorts in 200817. DVWA is a 276 amino acid protein that binds to β-tubulin, modulating its chondrogenic function18. The gene was also found to be more highly expressed in cartilage than in other human tissues, suggesting an involvement of DVWA in the metabolism of cartilage in humans (Table 1).
A further GWAS for knee OA in Japanese participants identified association with further SNP rs7775228 (OR 1.34, 95% CI 1.21–1.49, p = 2.43 × 10−8) and rs10947262 (OR 1.32, 95% CI 1.19–1.46; p = 6.73 × 10−8) in a region containing HLA class II/III genes (HLA-DQB1 and BTNL2), and was replicated in Europeans19, indicating that immunologic mechanisms may also contribute to OA pathogenesis.
The Treat-OA consortium GWAS reported and replicated an association of rs3815148 in COG5 on chromosome 7q22 with knee and hand OA in Europeans (p = 8 × 10−8)20. SNP in COG5 were in strong linkage disequilibrium (LD) with variants in 5 further neighboring genes (PRKAR2B, HPB1, GPR22, DUS4L, and BCAP29), complicating the identification of the functionally relevant genes. Gpr22 expression was absent in mice without OA, but present in mice with OA20. In a large metaanalysis and replication study, an SNP (rs4730250) in DUS4L was associated with knee OA at a genome-wide significant level (p = 9.2 × 10−9), indicating that any of these highly linked genes may contribute to the risk of developing knee OA21. Validated expression of these genes in the joint environment was also confirmed by functional analysis and gene expression studies using cartilage tissues from OA cases and controls, suggesting that HBP1 could be prioritized as a potential biomarker22 (Table 1).
The DOT1L gene was initially associated with the hip OA endophenotype joint space width (p = 1.1 × 10−11)23. Functional analysis in mice identified a role for Dot1L in chondrogenesis23. The same locus was previously associated with height and skeletal development22. A GWAS using individuals of European origin identified an association of rs12982744 with hip OA in men (p = 7.8 × 10−9, OR 1.17, 95% CI 1.11–1.23)24.
An additional SNP (rs6094710) situated near NCOA3 was established by a large metaanalysis of hip OA GWAS (OR 1.28, 95% CI 1.18–1.39, p = 7.9 × 10−9)25. NCOA3 demonstrates reduced expression in OA-affected articular cartilage when compared with macroscopically unaffected cartilage of the same joint. The involvement of NCOA3 expression in the setting of OA remains unclear and needs to be investigated, although involvement in hormonal regulation of bone turnover, such as thyroid hormones and steroids, or the suggested involvement of the gene in chondrocyte mechanotransduction are possibilities22 (Table 1).
The arthritis research council Osteoarthritis GENetics (arcOGEN) consortium carried out a 2-stage GWAS, culminating in the largest GWAS in OA to date. The initial hip and knee OA GWAS (sample size: 3177 cases and 4894 controls) and replication performed by the arcOGEN study in the United Kingdom did not identify any loci at genome-wide significance level, underlining the need for larger sample sizes and stricter phenotype definitions of OA given the complexity and heterogeneity of the disease7.
Following imputation of the initial GWAS and further metaanalysis, an SNP (rs11842874) in MCF2L was associated with knee OA at genome-wide significance (OR 1.17, 95% CI 1.11–1.23, risk allele frequency 0.93, p = 2.1 × 10−8)26. MCF2L has been reported to be involved in cell motility of the nervous system, indicating that the gene affects nociception22. Functional studies in zebrafish concluded that expression of MCF2L is involved in skeletal system development27 (Table 1).
The final larger-scaled arcOGEN GWAS identified the association of 5 novel loci with OA at genome-wide significance level and 3 additional novel loci at borderline significance. Almost 80% of the OA cases studied had undergone hip and/or knee arthroplasty, contributing to a severe OA phenotype definition28.
The strongest 2 signals established by this study were rs6976 (GLT8D1) and rs11177 (located in GNL3), both on chromosome 3p21.1 and in almost perfect LD with each other (OR for both SNP was 1.12, 95% CI 1.08–1.16, rs6976 p = 7.2 × 10−11, and rs11177 p = 1.3 × 10−10). These 2 loci are associated with both hip and knee OA in both men and women. The association was stronger in subjects with total joint replacement (TJR), illustrating the need to use stricter phenotype OA definitions. Further functional analysis is needed to unravel the underlying mechanisms28 (Table 1).
An intronic SNP (rs4836732) situated in ASTN2 was associated with hip OA in women who had undergone arthroplasty (OR 1.20, 95% CI 1.13–1.27, p = 6.11 × 10−10), also pointing out that stricter phenotype definition can empower analysis28. ASTN2 has been suggested to be involved in the regulation of the neuronal protein ASTN1, but the contributory involvement to OA mechanisms needs to be investigated (Table 1).
A further intronic SNP, rs835487 located in the CHST11 gene, was found to be associated with hip OA in TJR subjects with an OR of 1.13 (95% CI 1.09–1.18, p = 1.64 × 10−8) for both sexes combined28. CHST11 is differentially expressed in OA-affected and unaffected cartilage, and the gene is implicated in cartilage development. A further signal at rs9350591 was located between FILIP1 and SENP6. The signal reached genome-wide significance for hip OA (OR 1.18, 95% CI 1.12–1.25, p = 2.42 × 10−9)28. Although there is no suggested related function of FILIP1 or SENP6 in OA etiology yet, COL12A1 resides nearby and is known to be involved in bone formation29. An additional signal rs10492367, which lies between KLHDC5 and PTHLH, was also established as a risk locus for hip OA (OR 1.14, 95% CI 1.09–1.20, p = 1.48 × 10−8)28. PTHRP has been reported to be involved in mouse skeletal development30 (Table 1).
Three further variants were identified as associated with OA, but narrowly missed genome-wide significance in the arcOGEN study: the intronic variant rs8044769, located in the fat mass- and obesity-associated FTO gene, more highly associated in women; the intronic variant rs12107036 in TP63, more highly associated in women who had undergone TJR; and rs10948172, which lies between SUPT3H and CDC5L, more highly associated in men28. FTO is an established obesity risk locus31. The phenotypic overlap between OA and obesity has been genetically substantiated32, and the involvement of FTO in OA has been shown to be mediated through obesity33. The functional involvement of TP63 in the pathogenesis of OA is unclear, although it is reported to be involved in facial shape development34. The functional involvement of SUPT3H and CDC5L remain undefined in OA, although RUNX2, a closely located gene in extended LD, plays a regulatory role in bone development35 (Table 1).
Success of genetic locus detection in OA versus the inflammatory arthritides
In contrast to OA, the primary signal in the autoimmune arthritides, such as rheumatoid arthritis (RA) and juvenile idiopathic arthritis, resides in the MHC at the HLA loci. Numerous further loci genome-wide have been identified, many of which overlap across rheumatic diseases but not with them36. The main reasons behind these differences are thought to be the different etiopathology of OA, increased heterogeneity of the condition, and the relatively smaller GWAS conducted to date. For example, the largest metaanalysis of RA GWAS to date reached 29.9K individuals with disease compared with 7.5K patients with OA in the largest OA GWAS to date37.
Epigenetics of OA
There is existing evidence that implicates the involvement of epigenetic gene expression regulatory mechanisms in the pathophysiology of OA. Several genome-wide methylation studies for hip and knee OA have been conducted to date and most focus on the cartilage38,39,40,41,42. These studies demonstrate differentially methylated regions that include genes that have been associated with OA and distinct clustering of hip and knee samples. The concordance of these studies’ results is limited. Further epigenetic research could enable understanding of cartilage engineering mechanisms and lead to novel epigenetic-based therapeutic strategies for OA treatment.
FUTURE PROSPECTS
OA phenotypic definition and endophenotypes
The site- and sex-specific heterogeneity of OA is reflected in the associated genetic heterogeneity. Of the 18 established OA susceptibility loci, only 1 has been associated with both knee and hand OA, 5 with both knee and hip OA, 7 with hip OA only, and 6 with knee OA only (Table 1). Similarly, 13 loci have been associated with OA in both male and female subjects, 3 in women only, and 2 in men only. The examination of narrower, more homogeneous phenotypic definitions of OA such as disease severity (e.g., arthroplasty), combined with radiographic severity grading and disability measurements, specific morphological variations, anatomic differences of patterns of the affected joints, and stratification by sex are expected to further empower the identification of the genetic determinants of OA. Bone geometry is also thought to be genetically determined and may be a contributory factor43,44. Pistol grip deformity (femoral head shape) and neck shaft angle have been associated with the hip OA phenotype45. There is also an association between joint deformities quantified by measuring morphologic indices including alpha angle, triangular index height, lateral center edge angle, and extrusion index on pelvic radiographs with the risk of hip arthroplasty and an endstage OA phenotype46. These findings underline the importance of further exploration of the genetic basis of well-defined OA-associated endophenotypes. For example, the OA-associated locus in the DOTL1 gene was first discovered through its association with cartilage thickness (measured by joint space width on radiographs) in a relatively small population23. The outcomes from the arcOGEN study also suggest that use of precise endophenotypes has the potential to enhance the detection of relevant associations28.
Technological and analytical advances
In keeping with other common and complex diseases, the total of 18 OA-associated variants in Asian and European populations are represented by common SNP with moderate to small effect sizes. Some of the established signals do not replicate in all ethnic groups tested so far, even when the power of the replication cohorts is sufficiently high. These findings explain only a modest fraction of the heritable component of OA (Figure 1), although the application of genome-wide methods to assess the proportion of genetic variance explained by SNP is expected to increase this estimate8.
The use of high-throughput genotyping and sequencing approaches along with more advanced imputation (for example, using the Haplotype Reference Consortium data: www.haplotype-reference-consortium.org) will enable the interrogation and identification of variants from the full allelic frequency spectrum. Low frequency (MAF 1–5%) and rare variants (MAF < 1%) may have larger effect sizes and point to more functionally relevant regions and causal genes. The bigger the biological effect a gene has, the more likely it is to provide important clues about the underlying disease process. There are several examples of rare variants contributing to complex disease47 and it is likely that OA susceptibility will also be underpinned by variants across the MAF spectrum. In addition, the focus of OA genetics studies to date has been on biallelic sequence changes. It is likely that copy number variation may also be involved. The decreasing cost of high-depth whole genome sequencing is poised to make high-throughput, large-scale genetic studies possible in the near future.
Integrative analysis
Systematic functional genomics approaches in relevant tissues will help characterize the molecular landscape of OA alongside genetics approaches. To understand and treat this multifunctional and complex chronic disease, analytical approaches need to model disease biology from a holistic perspective48. Integrative data analysis including targeted approaches such as transcriptomics, proteomics, metabolomics, and methylomics can help in this regard. These large heterogeneous datasets require integration and support by advanced software methodology and novel analysis tools49. This approach necessitates multidisciplinary research50, although studies using integrated molecular genome-wide profiling of relevant tissue have thus far been limited. Integrating the range of different approaches in OA research is the next step to unravel the underlying mechanisms of the whole joint failure.
Over the last few years there has been a profound transformation in the ability to identify genes that contribute to musculoskeletal disorders. The OA susceptibility loci established to date point to several biological pathways, but further functional analysis is needed to identify the causal genes and determine their involvement. Identification of the missing heritability could be enhanced through the use of advanced genotyping technologies, imputation, stricter OA phenotypic definitions, and the establishment of larger and better-powered studies and metacollaborations. Systematic integrative analysis is also expected to lead to a profound transformation in the ability to holistically understand this complex disease. The future opportunities of OA genetic research are brightened by all these novel approaches.
Acknowledgment
We are grateful to Eleftheria Zeggini for commenting on the manuscript.
- Accepted for publication September 23, 2015.








{kind=link}