Article Text

Abstract
Objectives Tumour necrosis factor (TNF) receptor-associated periodic syndrome (TRAPS) is caused by TNFRSF1A mutations, known to induce intracellular retention of the TNFα receptor 1 (TNFR1) protein, defective TNFα-induced apoptosis, and production of reactive oxygen species. As downregulation of autophagy, the main cellular pathway involved in insoluble aggregate elimination, has been observed to increase the inflammatory response, we investigated whether it plays a role in TRAPS pathogenesis.
Methods The possible link between TNFRSF1A mutations and inflammation in TRAPS was studied in HEK-293T cells, transfected with expression constructs for wild-type and mutant TNFR1 proteins, and in monocytes derived from patients with TRAPS, by investigating autophagy function, NF-κB activation and interleukin (IL)-1β secretion.
Results We found that autophagy is responsible for clearance of wild-type TNFR1, but when TNFR1 is mutated, the autophagy process is defective, probably accounting for mutant TNFR1 accumulation as well as TRAPS-associated induction of NF-κB activity and excessive IL-1β secretion, leading to chronic inflammation. Autophagy inhibition due to TNFR1 mutant proteins can be reversed, as demonstrated by the effects of the antibiotic geldanamycin, which was found to rescue the membrane localisation of mutant TNFR1 proteins, reduce their accumulation and counteract the increased inflammation by decreasing IL-1β secretion.
Conclusions Autophagy appears to be an important mechanism in the pathogenesis of TRAPS, an observation that provides a rationale for the most effective therapy in this autoinflammatory disorder. Our findings also suggest that autophagy could be proposed as a novel therapeutic target for TRAPS and possibly other similar diseases.
- Cytokines
- Fever Syndromes
- Inflammation
Statistics from Altmetric.com
Introduction
Heterozygous mutations of the TNFRSF1A gene, encoding the 55 kDa tumour necrosis factor (TNF)α receptor 1 (TNFR1), have been detected in patients with TNF receptor-associated periodic syndrome (TRAPS; OMIM 142680), a periodic syndrome characterised by prolonged episodes of fever, multiorgan involvement and, in the most severe cases, systemic amyloidosis.1–5 TNFR1 mediates TNFα signalling, and ligand–receptor binding induces two alternative pathways, leading to either NF-κB activation or apoptosis.6 ,7
The molecular pathogenesis of TRAPS is not yet completely understood, and defects in either TNFR1 intracellular trafficking, receptor shedding or induction of apoptosis have so far been hypothesised.8–13 In particular, TNFRSF1A mutations have been observed to cause incorrect localisation of TNFR1 proteins, with retention in the endoplasmic reticulum (ER) and accumulation due to impaired protein elimination, in both in vitro and in vivo models and in leucocytes obtained from patients with TRAPS. As a consequence, elevated levels of reactive oxygen species (ROS) have been observed in immune cells from patients with TRAPS harbouring TNFRSF1A mutations,14 whereas neutrophils from these patients have shown impaired induction of apoptosis,9 defects that probably contribute to the typical prolonged inflammation status.
Inhibition of cytokines of innate immunity (mainly interleukin (IL)-1β and TNFa) signalling has turned out to be the most successful pharmacological approach,5 ,15 ,16 suggesting that the abnormal inflammatory response in TRAPS is mediated by increased secretion of proinflammatory cytokines.
Recent observations have shown a link between inflammation and autophagy, the main mechanism responsible for elimination of both damaged cellular compartments in physiological conditions and insoluble aggregates of mutant proteins that accumulate under pathological circumstances. In particular, downregulation of autophagy is known to trigger innate immune responses in cells, causing inflammation, such as in mice lacking autophagy-related proteins.17 ,18
As the above papers suggest a link between inflammasome and autophagy, in the present work we tested the hypothesis that an autophagy defect is a common pathogenic event underlying a number of autoinflammatory syndromes, including TRAPS.
Materials and methods
Patients
Ten Italian patients with TRAPS were involved in the study (see online supplementary table S1). Peripheral blood samples were collected from healthy donors and patients with TRAPS and processed to isolate monocytes, as described in online supplementary methods. The study was approved by the ethics board of ‘G Gaslini’ Institute, and informed consent was obtained from patients and controls.
Cell culture, transient transfections and pharmacological treatments
Human embryonic HEK-293T cells (abbreviated to ‘293T’) were grown and transfected under conditions reported in online supplementary methods, with one TNFRSF1A expression construct at a time, carrying either the wild-type (WT) or a mutant allele. Drugs were added to cells 1 day after transfection for a further 24 h (see online supplementary table S2).
Fluorescence microscopy analysis
293T cells were transfected with 500 ng expression constructs encoding the WT or mutant forms of TNFRSF1A and analysed after 48 h. Dots localising the LC3B autophagic protein were quantified by using the pEX-HcRed-hLC3WT plasmid (Addgene plasmid 24991),19 encoding LC3B fused to the HcRed fluorescent protein. Immunofluorescence studies were carried out as previously described20 (see online supplementary methods and supplementary table S3).
Cell lysates and western blot assay
293T cells were grown in 60 mm plates and transfected with 1 μg plasmids expressing TNFR1 proteins. Two days after transfection, total lysates in radio-immunoprecipitation assay (RIPA) buffer or soluble and insoluble fractions of 293T cells were obtained and processed (see online supplementary methods) as previously described,21 with antibodies reported in online supplementary table S4.
Cell stimulation and IL-1β secretion
Monocytes, isolated as described in online supplementary methods and by Gattorno et al,22 were seeded in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum (FBS) at 37°C, treated for 24 h with or without geldanamycin (GA) and stimulated for 6 h and 24 h with or without 1 μg/ml lipopolysaccharide (LPS) (Sigma).
Results
TNFRSF1A mutations lead to intracellular inclusions
In accordance with previously described in vitro and ex vivo experiments,10 ,23 fluorescence microscopy analysis revealed that WT TNFR1 protein was located both inside the cytosol and on the plasma membrane (figure 1A, left panel), whereas mutant proteins were located mainly in intracellular inclusions represented by small (<2 μm) and large (>2 μm) aggregates (figure 1A, right panel and bars diagram).

Cellular localisation of tumour necrosis factor (TNF)α receptor (TNFR) proteins. (A) Fluorescence analysis of 293T cells expressing green fluorescent protein-fused TNFR1 constructs shows large aggregates, typically produced by mutant proteins. Percentages of cells containing aggregates are the mean±SD of three independent experiments; differences refer to cells expressing the wild-type (WT) protein (unpaired Student t test: *p=0.02; **p=0.002). (B) Fluorescence-activated cell sorter analysis of 293T cells, expressing WT and mutant TNFR1 proteins, all showing comparable transfection efficiency, labelled with a TNFR1-specific antibody without cell permeabilisation. Therefore the blue peaks represent the percentage of TNFR1 protein detected on the cell surface of 293T cells by the specific antibody against human TNFR1 compared with isotype control (red peaks). Results from three independent experiments are reported as percentage of positive cells.
Quantitative evaluation of cell surface TNFR1 protein showed significantly reduced expression only on 293T cells transfected with mutant C55Y and C43R constructs. In contrast, TNFR1 cell surface expression in cells carrying the rare TRAPS-associated mutation, del27 (c.586–612; DEL27), or the clinically debated R92Q mutation was similar to the WT protein (figure 1B). This experiment further confirms that TRAPS-associated mutations affecting cysteine residues have the strongest impact on subcellular localisation of TNFR1 proteins.10 ,11 ,23 ,24
Intracellular accumulation of the TNFR1 mutant proteins leading to insoluble aggregates13 was also confirmed by a different distribution of the WT and mutant TNFR1 proteins in the soluble and insoluble fractions of transfected 293T cells (see online supplementary figure S1A), as well as by immunofluorescence carried out in monocytes and neutrophils derived from a healthy individual (Ctrl) and two patients with TRAPS carrying the C55Y and C43R TNFRSF1A mutations (see online supplementary figures S1B,C), supporting the suitability of our cellular model for further investigating TRAPS pathogenesis.
Autophagy is involved in TNFR1 protein degradation
As mechanisms underlying accumulation of TNFR1 mutant proteins seem to rely on a defect in protein degradation and clearance rather than excessive protein production,13 cells transfected with either WT or mutant (C55Y or DEL27) TNFR1–GFP (green fluorescent protein) tagged constructs were incubated with the proteasome inhibitor, lactacystin, or the autophagy inhibitor, 3-methyladenine (3-MA), to evaluate involvement of these processes in TNFR1 clearance.25 ,26 As shown in figure 2, treatment with lactacystin caused a marked reduction in both WT and mutant TNFR1 proteins, while the use of 3-MA induced a marked accumulation of all TNFR1 proteins, suggesting that autophagy is the main cellular mechanism for the clearance of TNFR1 proteins. Proteasome inhibition has been reported to induce autophagy in several cellular models,27 and this may explain the increased levels of the autophagosome marker, LC3BII, observed after treatment with proteasome inhibitors in 293T cells transfected with TNFRSF1A constructs (figure 2B). Moreover, ubiquitin proteasome system impairment, either constitutive or induced by TNFR1 mutant proteins, was excluded in our 293T cell system by using the plasmid, ZsProSensor-1, which expresses the proteasome-sensitive fluorescent ZsGreen (figure 2C,D).

Cellular mechanisms of tumour necrosis factor (TNF)α receptor 1 (TNFR1) protein elimination. (A) Flow cytometry analysis of TNFR1–GFP in 293T cells untreated (untr, white) or treated with either the proteasome inhibitor, lactacystin (grey), or the autophagy inhibitor, 3-methyladenine (3-MA, black). Results from three independent experiments are shown as mean fluorescence intensity (MFI) and reported as percentage ± SD with respect to untreated samples (unpaired Student t test: *p = 0,05; **p = 0.02). (B) Western blot analysis of total lysates from 293T cells, transfected with constructs encoding the wild-type (WT) or the C55Y proteins without any treatment (–) or in the presence of a proteasome inhibition (lactacystin). The amount of the housekeeping protein, tubulin, in each sample is shown in the bottom line. (C) Fluorescence microscope evaluation of the proteasome-sensitive ZsGreen protein in 293T cells transfected with the ZsProSensor-1 plasmid, without any treatment (untr) or in the presence of proteasome inhibition (MG132); images were obtained by using two different lens (10× and 100×). Nuclei are counterstained with DAPI (4',6-diamidino-2-phenylindole). (D) Western blot analysis of total lysates from HEK293 cell line stably expressing the proteasome-sensitive ZsGreen protein and transfected with pcDNA3.1TOPO/CT/GFP (empty) vector or with expression constructs for WT and mutant TNFR1 proteins. MG132 treatment, shown in the far right lane, was used as a positive control for ZsGreen expression. The membrane was labelled with ZsGreen, GFP and tubulin-specific antibodies.
Effect of mutant TNFR1 proteins on autophagy
Defective clearance of misfolded proteins may lead to further accumulation of intracellular proteins and subsequent inflammation, as has been observed with mutant cystic fibrosis transmembrane conductance regulator (CFTR) proteins.28 Moreover, animal models suggest that autophagy may be a common pathogenic event underlying a number of autoinflammatory diseases17 such as TRAPS.
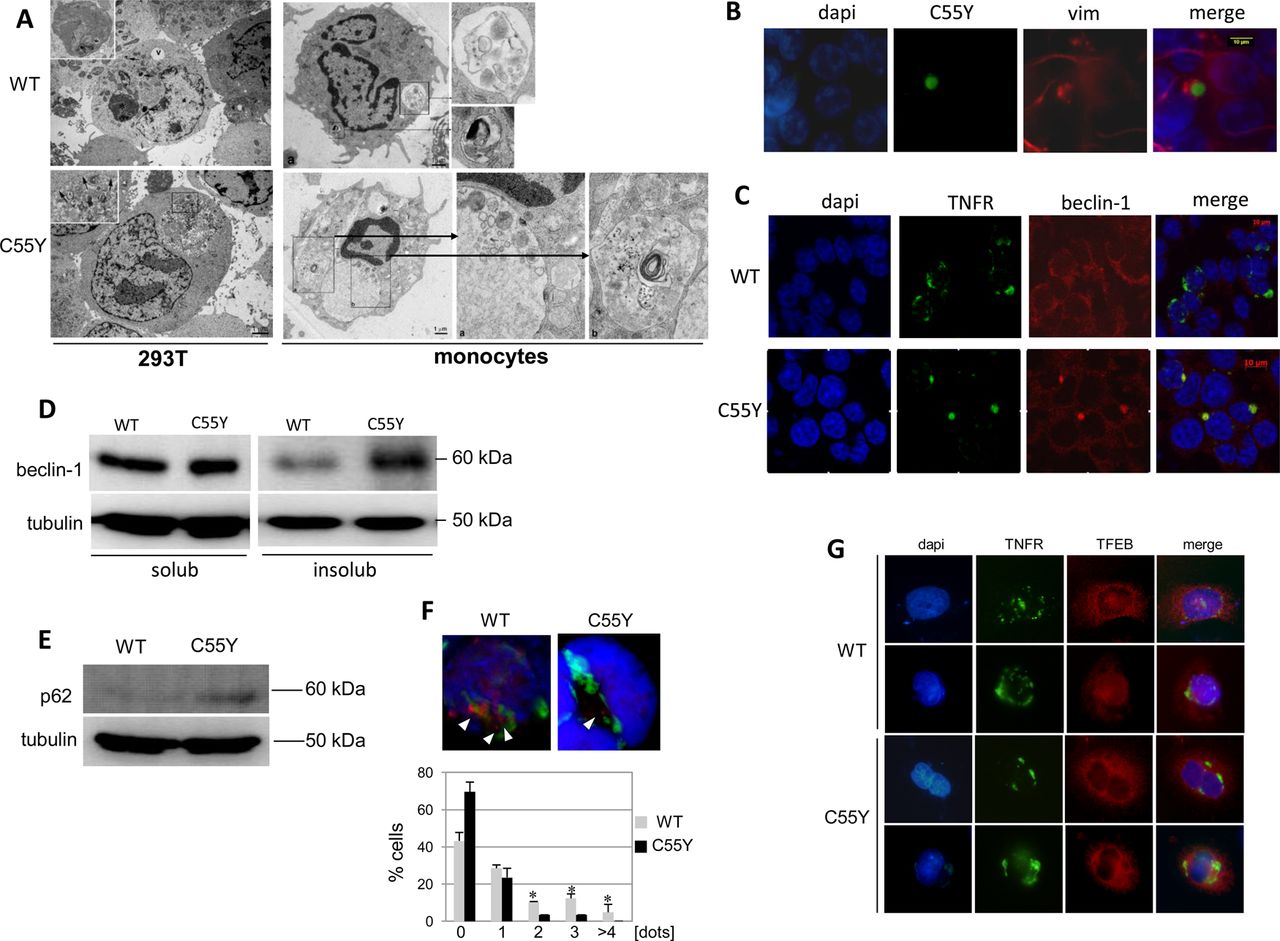
Consistent with this hypothesis, 293T cells expressing mutant C55Y TNFR1 and monocytes from patients with TRAPS carrying the C55Y mutation showed similar abnormalities on electron microscopy (figure 3A). Indeed, while monocytes from normal subjects displayed a normal complement of organelles, lysosomes and autophagosomes, cells obtained from the patient with TRAPS showed multiple, abnormally large autophagic vacuoles containing organelle remnants featuring ER vesicles and Golgi cisternae, ribosomes and glycogen particles, in addition to cytoplasmic sequestration by ER membranes. Taken together, the ultrastructural findings suggest that, in patients’ monocytes, initiation of the autophagic process occurs normally, but lysosomal digestion of the sequestered organelles is substantially impaired, leading to accumulation of enlarged autophagic vacuoles within the cells.
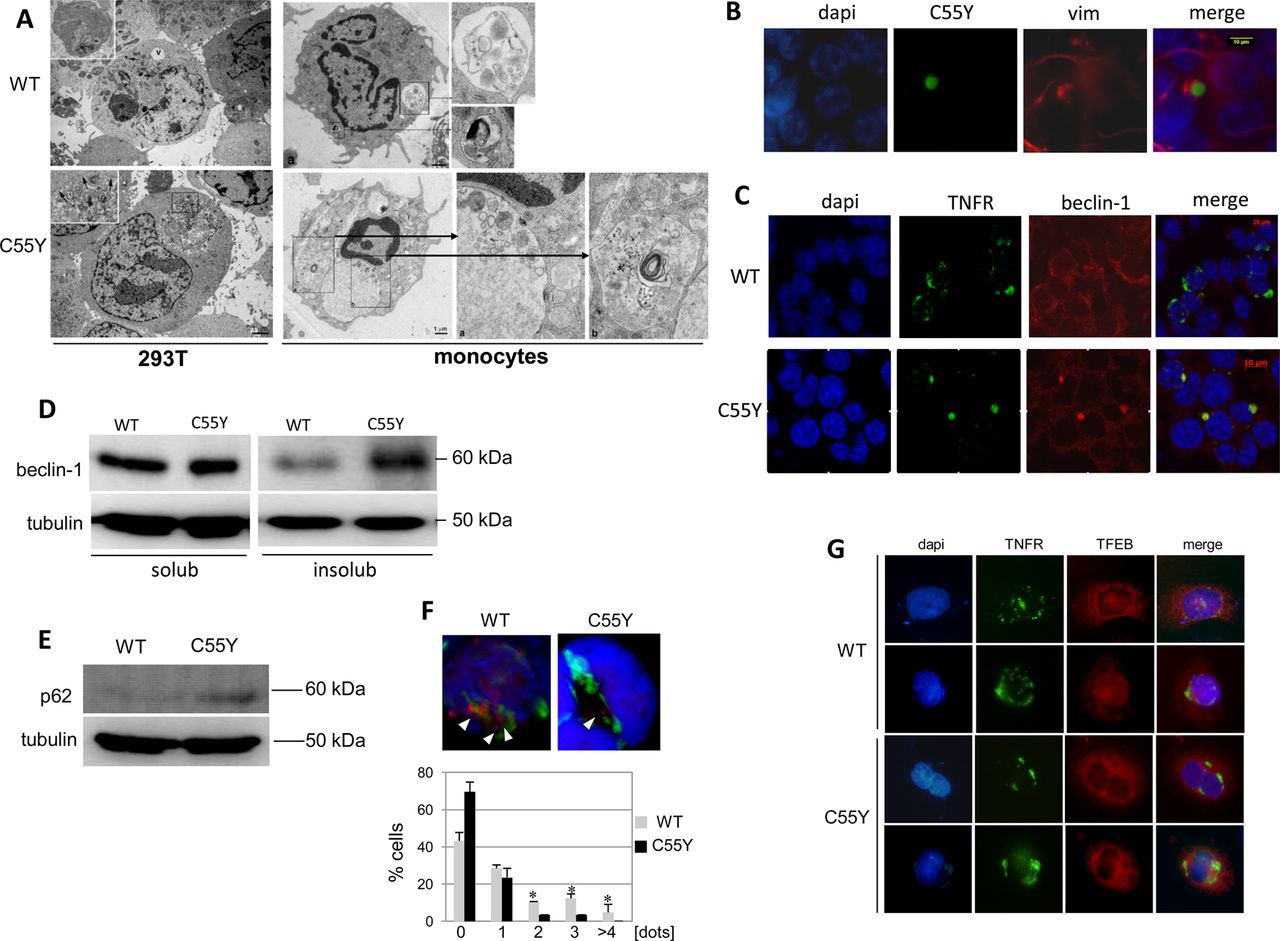
Autophagy in monocytes and 293T cells expressing tumour necrosis factor (TNF)α receptor 1 (TNFR1) proteins. (A) Electron microscopy analysis of 293T cells expressing wild-type (WT) (one sample) or C55Y (two replicated samples) TNFR1 proteins (left pictures). A large autophagic vacuole (indicated as ‘v’) and an autophagosome containing partially indigested material are enlarged in the upper left image. Arrows in the lower image indicate small lysosomes. Monocytes from a healthy individual (WT) and a patient with TNF-receptor associated periodic syndrome (C55Y) analysed at the moment of disease flare are shown in the right pictures. Two autophagic vacuoles (enlarged boxes) containing structures under active digestion (upper image), and two vesicles characterised by cytoplasmic indigested material (enlarged at the right side of the bottom image) are shown. (B) Fluorescence microscopic analysis of vimentin in 293T cells expressing the C55Y mutant protein. Original magnification 100×. (C) Fluorescence microscopic analysis of beclin-1 in 293T cells expressing the WT or C55Y proteins. Original magnification ×100. (D) Western blot analysis of beclin-1 amount in both soluble and insoluble fractions of lysates obtained from 293T cells transfected with constructs encoding the WT or mutant C55Y proteins. The housekeeping protein, tubulin, is shown in the bottom line. (E) Western blot analysis of p62 in total lysates from 293T cells transfected with WT or C55Y constructs. The housekeeping protein, tubulin, is shown in the bottom line. (F) Fluorescence microscopy analysis of 293T cells co-transfected with a LC3B–HcRed fusion and either WT or C55Y TNFR1–GFP constructs; the active form, LC3BII, is represented by red dots (see arrows). Below, the percentage of cells expressing TNFRSF1A WT or C55Y and characterised by 0, 1, 2, 3, 4 or more punctuate red dots is reported in the bars diagram as the mean±SD of three independent experiments. To assess significant differences in the number of dots between WT and C55Y, we carried out an unpaired Student t test (*p≤0.05). (G) Fluorescence microscopy analysis of the transcription factor EB (TFEB) cellular localisation, under rapamycin-mediated autophagy induction, in HeLa cells stably expressing the TFEB–Flag protein (red) and transfected with the expression constructs encoding WT TNFR1 (upper lines) or the mutant C55Y protein (bottom lines) fused to the GFP protein (green). For each TNFR1 construct tested, two cells are represented. Unfortunately, the low transfection efficiency of TNFRSF1A plasmids in cells stably expressing TFEB did not allow us to perform quantitative evaluations of TFEB nuclear translocation.
Because formation of aggresomes, reflected by delocalisation of vimentin which surrounds the mutant C55Y TNFR1 protein aggregates (figure 3B), suggested a defect in protein elimination, we made additional observations consistent with the hypothesis of mutant TNFR1 inducing autophagy impairment28: (i) retention of beclin-1 within both aggregates containing the mutant protein (figure 3C) and the insoluble fraction (figure 3D); (ii) accumulation of p62 (figure 3E); (iii) decreased number of LC3BII dots (figure 3F); (iv) decreased nuclear translocation of the master gene of lysosomal biogenesis, transcription factor EB (TFEB) (figure 3G). The gene expression of beclin-1, LC3B and p62 was not affected by mutant TNFR1 proteins (real-time PCR, data not shown), further corroborating our hypothesis of a direct effect of mutant TNFR1 on autophagy-mediated protein elimination. Moreover, real-time PCR showed that expression of the lysosomal gene, LAMP1, a TFEB target, was more strongly induced in the presence of WT TNFR1 than in the presence of C55Y mutant protein (not shown).
Effects of GA on TNFR1 localisation
HSP90 inhibition by the antibiotic GA has been reported to induce protein refolding in cellular models of several diseases.20 ,29 ,30 Moreover, as HSP90 is involved also in the regulation of NF-κB activity, and GA-mediated HSP90 inhibition leads to IκB kinase (IKK) subunit degradation,31 we investigated the effects of GA in terms of both trafficking of mutant misfolded TNFR1 proteins and TNFR-dependent NF-κB activation.
Fluorescence microscopy analysis of transfected 293T cells showed that, while GA had no effect on degree of WT TNFR1 dispersal (figure 4A, upper panels), it could induce both formation of a diffuse distribution in cells expressing the C55Y protein (figure 4A, lower panels) and a significant reduction in large aggregates (figure 4A, bottom graph). As decreased aggregation rate may depend on protein refolding, protein elimination or both, cytofluorimetric analysis using a TNFR1-specific antibody without permeabilisation allowed us to assess a significant increase in the rescue of membrane localisation for C55Y, C43R and DEL27 mutant proteins (red line), with respect to untreated cells (blue line) (figure 4B, upper fluorescence-activated cell sorter images and lower diagram plot), while R92Q and WT proteins did not gain any further enrichment on the plasma membrane, suggesting that the GA refolding effect probably occurs only in the presence of mutant TNFR1 proteins (figure 4B and online supplementary figure S2A for internal isotypic controls). Rescue of the surface localisation of mutant TNFR1 was also achieved, upon GA treatment, on monocytes from four patients with TRAPS carrying cysteine mutations (see effect on one ‘C55Y’ in figure 4C), which reacted by increasing the trafficking of TNFR1 proteins to the cell membrane by 2.8-fold, 4.1-fold, 3.8-fold and 2.5-fold. On the other hand, monocytes from two healthy individuals did not show any significant GA-mediated TNFR1 membrane increase (figure 4C and online supplementary figure S2B for internal isotypic controls).
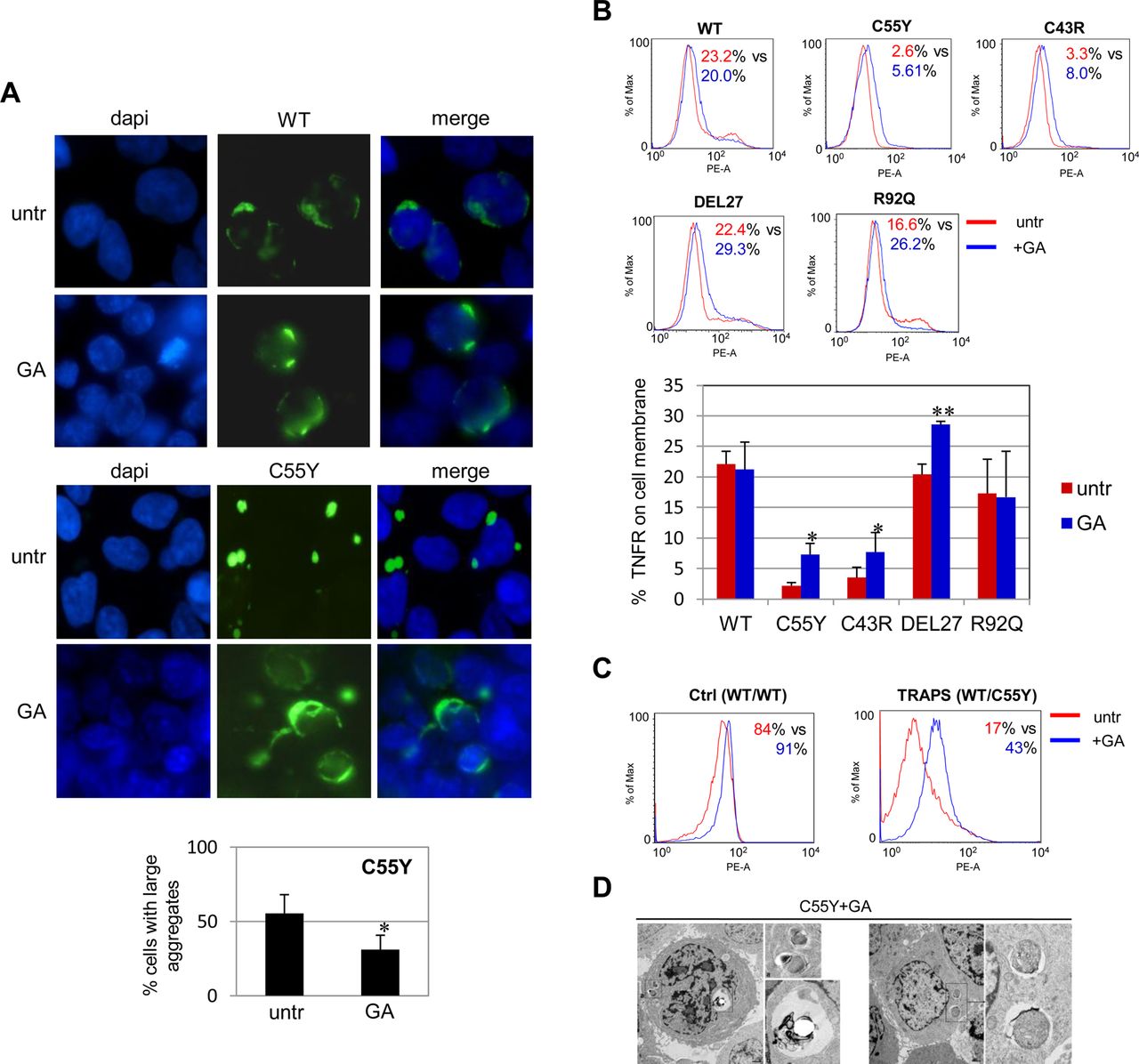
Effect of geldanamycin (GA) on cellular localisation of tumour necrosis factor (TNF)α receptor 1 (TNFR1) proteins. (A) Fluorescence microscopy analysis of 293T cells transfected with the wild-type (WT) (top panels) and C55Y (bottom panels) TNFR1 constructs either untreated (untr) or treated with GA 360 nM for 24 h. The percentage of cells transfected with the mutant TNFR1 with or without GA, and containing large aggregates is reported in the diagram below and represents the mean±SD of three independent experiments (paired Student t test: *p<0.05). Original magnification ×100. (B) Flow cytometry analysis in 293T cells transfected with WT, C55Y, C43R, DEL27 or R92Q TNFR1–GFP constructs, without any treatment (red, untr) or treated with GA 360 nM (blue, GA), labelled with a TNFR-specific antibody without cell permeabilisation. Results reported in the diagram below are the mean±SD of three independent experiments. Asterisks indicate significant differences between untreated and treated cells in terms of mean fluorescence intensity quantification of the rescue of membrane localisation (paired Student t test: *p<0.05; **p<0.01). (C) Flow cytometry analysis of TNFR1 in monocytes from a healthy individual (ctrl, WT/WT) and a C55Y patient (WT/C55Y), either untreated (red) or treated with GA 360 nM for 24 h (blue). The percentage of positive cells before versus after treatment is also reported. (D) Electron microscopic evaluation of 293T cells transfected with the C55Y construct and treated with GA 360 nM for 24 h (two panels).
We also observed, by directly detecting GFP fluorescence, a significant reduction in total C55Y mutant protein after GA treatment, compared with WT, suggesting that the GA-mediated refolding effect acts in concert with mechanisms of elimination of the C55Y-TNFR1 protein20 ,30 (see online supplementary figure S2C). The amount of mutant C55Y-TNFR1 protein was restored to high levels after combined treatment with GA and the autophagy inhibitor, 3-MA, confirming that GA-induced TNFR1 elimination is mediated by autophagy effects (see online supplementary figure S2D). Indeed, autophagy has turned out to be directly induced by GA treatment, as demonstrated by increased levels of the autophagosome marker, LC3BII, after GA treatment in 293T cells (see online supplementary figure S2E), an effect that was also detected in the THP-1 monocytic cell line (not shown).
Therefore GA seems able to counteract the deleterious consequences of TNFR1 mutations, as further demonstrated by the rescue of a normal ultramicroscopic phenotype in GA-treated 293T cells expressing the C55Y protein (figure 4D).
Effect of GA on TNFR-mediated NF-κB activation
NF-κB is crucial in inflammatory responses, and activated in cells derived from patients with TRAPS.23 ,32 ,33 The balance between HSP90 and HSP70 affects NF-κB by modulating, among other steps, IKK subunit degradation.34 As HSP90 takes part in IKK complex turnover, and is negatively regulated by GA, we investigated whether this antibiotic could also exert an effect on TNFR1-induced NF-κB activation.
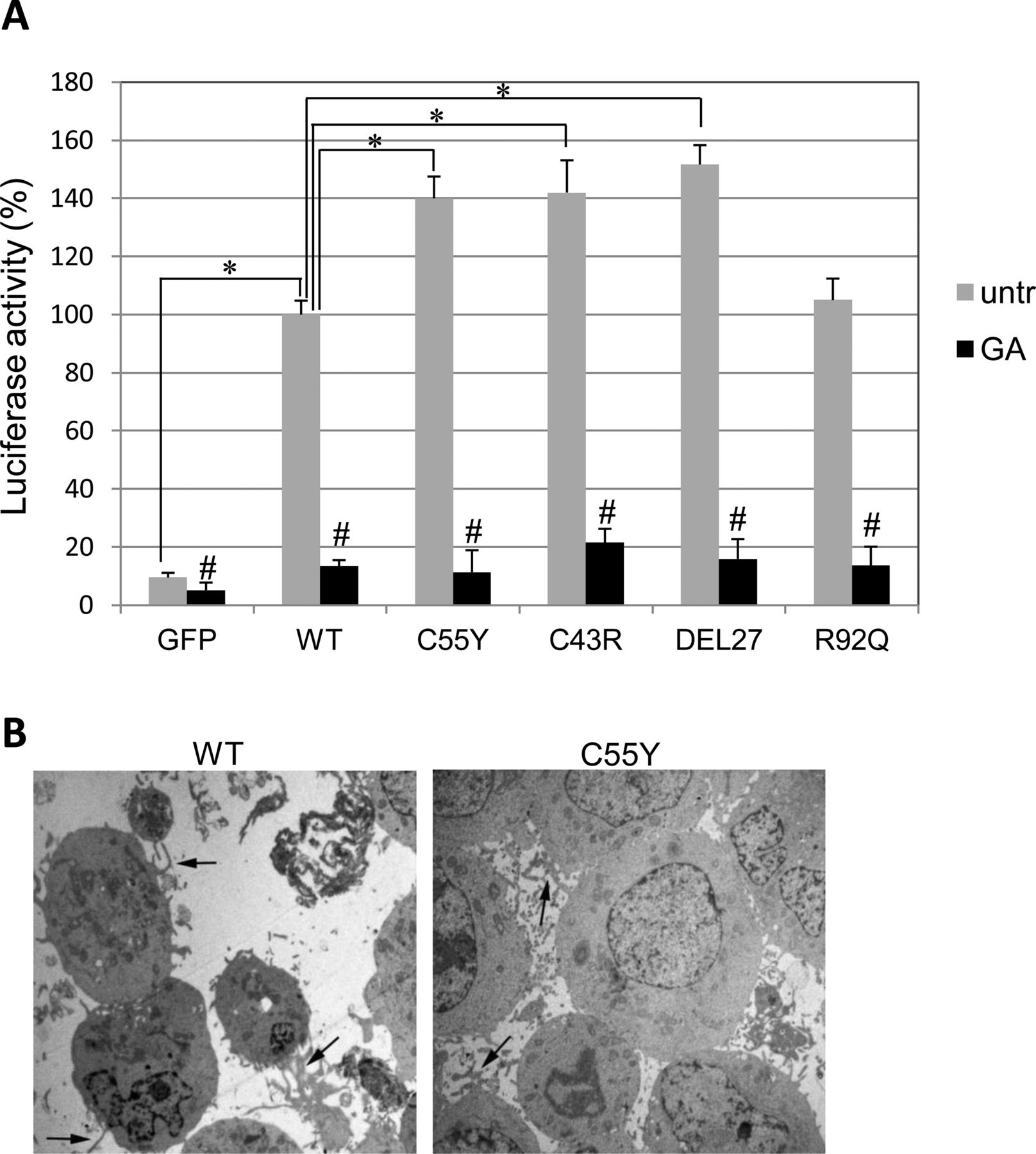
Luciferase activity driven by NF-κB binding elements cloned upstream of a Luciferase reporter gene showed that mutant TNFR1 proteins did trigger NF-κB activation greater than the WT protein (figure 5A, grey bars), consistent with TRAPS-associated abnormal inflammation.32 ,33 This observation was further confirmed by electron microscopy, which showed 293T cells expressing C55Y-TNFR1, but not WT-TNFR1, associated with a large number of lamellipods and pseudopods, markers of excessive cellular activation (figure 5B).

Effect of tumour necrosis factor (TNF)α receptor (TNFR) mutations on NF-κB activity. (A) Luciferase analysis in 293T cells co-transfected with TNFR1 or empty GFP constructs and a reporter plasmid containing five NF-κB recognition sequences upstream of the Luciferase gene, in the absence (grey bars) or presence (black bars) of geldanamycin (GA). Results are expressed as percentage of the wild-type (WT) untreated condition (referred to as ‘100’) and are the mean±SD of three independent experiments. Statistical comparisons included (i) untreated WT versus untreated mutant or GFP alone (unpaired Student t test: *p<0.05) and (ii) each untreated WT or mutant cell sample compared with the GA-treated cells (paired Student t test: #p<0.05). (B) Electron microscopic images of 293T cells transfected with WT or C55Y TNFRSF1A construct. Cells expressing the mutant TNFR1 show a higher amount of lamellipods and pseudopods, markers of activated cells (black arrows).
Moreover, after assessing the ability of GA to interfere with the NF-κB pathway, by inducing HSP70 overexpression and IKKγ partial elimination (see online supplementary figure S3A,B), and verifying that these effects occur also in the monocytic THP-1 cell line (not shown), we demonstrated that transfected 293T cells treated with GA for 24 h show a dramatic reduction in NF-κB activation, suggesting that the effects of mutant TNFR1 proteins on NF-κB can be reversed (figure 5A, black bars).
Effect of GA on IL-1β production by TRAPS monocytes
As the most effective treatments of TRAPS rely on counteracting inflammation by inhibiting intracellular IL-1β signalling,15 we investigated whether GA may also affect the IL-1β pathway in TRAPS monocytes. To this end, we assessed the level of IL-1β secretion after LPS stimulation in the absence or presence of GA in monocytes isolated from both healthy individuals and patients with TRAPS. A higher secretion of IL-1β after 6 and 18 h of LPS stimulation was observed in monocytes from inactive and untreated patients compared with both age-matched healthy controls and patients with inactive TRAPS under continuous treatment with IL-1 blockers (figure 6A). No difference between the three groups after 3 h of LPS stimulation was observed (not shown). GA was able to dramatically reduce IL-1β secretion in monocytes from all the groups (figure 6B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pattern of interleukin (IL)-1β secretion by monocytes from patients with tumour necrosis factor (TNF)-receptor associated periodic syndrome (TRAPS) and effect of geldanamycin (GA). (A) Relative secretion of IL-1β after 6 h (white bars) and 18 h (grey bars) of LPS stimulation in monocytes from untreated patients and patients treated with IL-1β blockers compared with age-matched healthy controls. Bars represent median values and whiskers represent SE. Four untreated patients (three carrying the T50M mutation and one the C52Y mutation) were analysed during attack-free intervals (Mann–Whitney U test: NS). Four patients (three with the C55Y mutation and one with the C52Y mutation) were receiving anti-IL-1β treatment, with complete control of clinical and biological manifestations (Mann–Whitney U test: *p=0.01). IL-1β secretion in treated patients was significantly higher than in untreated patients after 18 h of LPS (Mann–Whitney U test: p=0.029), while the comparison gave an almost significant difference after 6 h of LPS (Mann–Whitney U test: p=0.057). (B) Relative secretion of IL-1β after LPS+GA stimulation compared with LPS alone assessed in healthy controls (black bars), untreated TRAPS (grey bars) and anti-IL-1β-treated TRAPS (white bars) patients (Wilcoxon pair test: **p<0.001). (C) Present data and mechanisms already reported on TRAPS pathogenesis are combined in the proposed model. Protein aggregation induced by mutations in the TNFRSF1A gene may lead to defective autophagy due to either overloading (as reported here) or, indirectly, to mTOR activation consequent on reactive oxygen species (ROS)-mediated NF-κB activation.39 Moreover, both autophagy impairment and ROS production can increase expression of proinflammatory cytokines by two alternative mechanisms, namely NF-κB and JNK/MAPK40 activation. GA acts on different steps, resulting in rescue of the autophagic function and therefore in a favourable phenotypic outcome.
Discussion
In this work, we report for the first time that TNFR1 proteins are cleared by autophagy, and that autophagy is impaired in the presence of mutant TNFR1 proteins. Indeed, mutant misfolded receptors can induce formation of toxic intracellular aggregates which, interfering with their autophagy-mediated degradation, drive TRAPS pathogenesis.
Consistent with previous observations,11 ,13 we have demonstrated that failure of mutant TNFR1 to be localised at the plasma membrane is associated with protein accumulation, both in vitro and in peripheral blood mononuclear cells derived from patients. Our subsequent investigation, aimed at understanding how misfolded TNFR1 proteins are eliminated, revealed that autophagy is the only mechanism effective in the clearance of TNFR1 aggregates and that it is probably overwhelmed by massive protein aggregation, such as that occurring in cells expressing critical levels of mutant TNFR1.
Autophagy impairment has already been found to be responsible for accumulation of misfolded proteins in cystic fibrosis, Huntington disease and Alzheimer's disease28 ,35 ,36 and, similarly to that observed in Alzheimer's disease brain,37 cells harbouring TNFRSF1A mutations show accumulation of autolysosomes filled with undigested or partially digested substrates. Moreover, LC3B dot quantification, beclin-1 entrapment and decreased nuclear TFEB translocation-expressing cells38 further demonstrated crucial, although not severe, autophagy impairment, as (i) cell viability was not affected and (ii) only slight accumulation of p62, a protein exclusively eliminated by autophagy, could be demonstrated.
According to the present results and literature data, a model that links mutations in TNFRSF1A, defective apoptosis and efficacy of anti-IL-1 treatments to impaired autophagy is schematically proposed in figure 6C. First, our observation that mutant TNFR1 proteins accumulate and trigger higher NF-κB activity than the WT receptor is consistent with (1) elevated ROS production found in blood cells of patients with TRAPS14 and (2) oxidative stress promoted by ligand-independent TNFR1 signalling.41 Since a direct effect of ROS on inflammasomes has been excluded,14 autophagy may account for the link between TNFR-induced ROS production and cytokine oversecretion.
NF-κB activity in TRAPS has long been debated and alternatively reported to be upregulated and downregulated.13 ,23 ,32 ,33 Of note, induction of the autophagy inhibitor, mammalian target of rapamycin (mTOR), by NF-κB activation has been observed to impair the autophagic process,39 suggesting that the NF-κB-dependent inhibition of autophagy may have consequences on inflammation when occurring in cells of the immune system. On the other hand, NF-κB hyperactivation in cells expressing mutant TNFR1 may be directly due to accumulation of both the autophagy-specific substrate, p62, reported to increase signals leading to cell survival,42 and IKK proteins, normally degraded by autophagy.43 Moreover, autophagy impairment has been reported to prevent TNFα-induced apoptosis,44 corroborating the relation between NF-κB and autophagy and providing an explanation for the defective apoptosis observed in patients with TRAPS.9
Furthermore, polymorphisms of the gene encoding the autophagic protein, ATG16L1, are associated with Crohn's disease,45 while Atg16L1 knockout mice have increased production of the IL-1β cytokine,17 the secretion of which is regulated by degradation of its precursor, pro-IL-1β, by autophagy.46 Indeed, increased secretion of IL-1β has been observed in monocytes isolated from untreated TRAPS patients compared with both treated patients and healthy controls, consistent with the beneficial effects of treatment with IL-1β blockers on symptom amelioration.15 The decrease in IL-1β secretion after GA treatment, in agreement with rescue of the autophagic process and prevention of aggregate formation, may also result from GA-mediated IL-1β gene downregulation, explaining why cells from healthy controls respond to GA.47
Overall, the involvement of autophagy in TRAPS pathogenesis provides a rationale for the apparent paradox that the most effective therapy for TRAPS is inhibition of the cascade signalling induced by IL-1β rather than the use of drugs that counteract the TNFR-mediated pathway, as one would expect.15 In addition to GA, other molecules, such as 17-N-Allylamino-17-demethoxygeldanamycin (17-AAG) and trehalose,48 are effective in inducing autophagy, suggesting that autophagy can be considered a novel target for TRAPS therapy. This notion is confirmed by a study reporting that IL-1β secretion is downregulated on activation of autophagy,49 and strengthened by impaired basal autophagy in autoinflammatory familial Mediterranean fever.50
Finally, cellular responses to GA and outcomes have been shown to depend on the kind of TNFRSF1A mutation; indeed, aggregates in cells expressing WT or TNFRSF1A mutations, the pathogenic role of which is still debated, were mostly unaffected by GA administration, as a large proportion of these proteins may not be pathogenic intracytoplasmic aggregates but either accumulate in the ER or are already located on the cell surface. This is the case for R92Q (found at an allele frequency of 4–5% in the control population, present in asymptomatic transmitting parents, and associated with a milder disease course and a high rate of spontaneous resolution16), which behaved similarly to WT in our experiments. Despite a marked intracellular aggregation of the mutant DEL27 protein but with relatively normal cell surface expression, a pathogenic effect of DEL27 is supported by the observation that GA treatment significantly increased the membrane localisation of the mutant protein. Such a heterogeneous pattern of cell responses might be ascribed to the unique nature of this mutation, which (i) has been reported so far in only one unusual medical case with a specific ethnic background,9 and (ii) affects a protein domain with no other mutation reported so far. These observations suggest that cysteine mutations are the most effective in inducing a dramatic cellular response, while the R92Q variant seems to be associated with weaker functional effects, confirming a mild role, if any, in TRAPS.
Taken together, our findings are consistent with the hypothesis that autophagy plays a crucial role in the physiological balance of TNFR1 proteins within cells: impairment of its efficiency due to protein overload may account for TRAPS pathogenesis through downstream effects leading to massive inflammation and severe apoptosis impairment, fully explaining the effectiveness of therapies that reduce IL-1β signalling.
Acknowledgments
We are grateful to Francesca Antonini (Core Facilities, Gaslini Institute, Genoa) for technical help. We are also indebted to Dr Anna Rubartelli (National Cancer Research Institute, Genoa) and Dr Elisabetta Traggiai (2nd Div Pediatrics, Gaslini Institute & Novartis Inst Biomedical Research, Basel) for helpful discussion. Financial support from Italian Telethon (grants GGP07236 and GGP09127) and the EU-EUROTRAPS project is gratefully acknowledged.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
MG and IC contributed equally to this article.
-
Contributors TB, SC, MG and IC designed the research; TB, SC, PC, DB, EDZ, AO and ADO performed the experiments; AF and AB provided tools and reagents; TB, SC, PC, DB, AO, MG and IC analysed results; TB, SC and MG wrote the manuscript; RR and AM discussed the data and edited the manuscript. All the authors edited the manuscript.
-
Competing interests None.
-
Ethics approval The ethics board of ‘G Gaslini’ Institute approved the study.
-
Provenance and peer review Not commissioned; externally peer reviewed.