Article Text

Abstract
Background Optimal treatment for rheumatoid arthritis (RA) after inadequate response (IR) to tumour necrosis factor α inhibitors (TNFi) remains uncertain.
Objective To compare the efficacy and safety of biological agents after TNFi-IR.
Methods A systematic literature search was carried out using Medline and Cochrane databases, as well as http://www.clinicaltrials.gov, and bibliographies of the retrieved literature were searched by hand. Randomised, placebo-controlled trials that enrolled patients with RA with TNFi-IR were included and American College of Rheumatology (ACR) response as primary efficacy outcome and adverse events (AEs), serious adverse events (SAEs) and serious infections (SIs) as safety measures were extracted. An indirect meta-analysis with pairwise comparisons of efficacy and safety data was then carried out using ORs or risk differences (RDs) in a random effects model.
Results In four randomised controlled trials with 24 weeks' follow-up, direct comparisons of abatacept, golimumab, rituximab and tocilizumab versus placebo showed statistically significant mean ORs of 3.3–8.9 for ACR20, 5.5–10.2 for ACR50 and 4.1–13.5 for ACR70. Risks of AEs, SAEs and SIs versus placebo were non-significant. Indirect pairwise comparisons of the four biological agents showed no significant differences in ACR50 and ACR70. Golimumab had a significantly lower OR (0.56–0.59) for ACR20 but significantly fewer AEs (RD 0.13–0.18). Efficacy after one versus multiple TNFi failures did not differ significantly between the different biological agents.
Conclusion In patients refractory to one or more TNFi, new biological agents provide significant improvement with good safety. Lacking head-to-head trials, indirect meta-analysis enables a comparison of effectiveness and safety of biological agents with each other and shows that all biological agents have similar effects.
Statistics from Altmetric.com
Introduction
In rheumatoid arthritis (RA), an inadequate response (IR) to initial synthetic disease-modifying antirheumatic drugs (DMARDs), such as methotrexate (MTX), leads to an escalation of treatment, often by addition of a tumour necrosis factor α inhibitor (TNFi). However, many patients fail to achieve a good response with a TNFi.1 In randomised controlled trials involving these inadequate responders (TNFi-IR), several biological agents have demonstrated efficacy versus placebo, but none of these drugs have been compared directly for efficacy or safety.
In the absence of head-to-head trials, indirect meta-analysis has emerged as an accepted and valid methodology for comparing drugs with each other using a common comparator—for example, placebo or a synthetic DMARD.2 A 2009 network meta-analysis3 provided a comprehensive examination of all biological agents in RA. The inclusion of all studies regardless of previous treatments, however, has elicited some uncertainty about the comparability of the included trial populations.4 To deal with this concern, we sought to identify the optimal biological agent for patients with RA after an inadequate therapeutic response to TNFi by examining drug efficacy and safety outcomes in randomised controlled trials (RCTs) limited to patients with TNFi-IR.
Materials and methods
Data sources and searches
We undertook a systematic literature search including RCTs that selected patients with RA for whom one or more biological treatment had previously failed. We searched the Medline and Cochrane databases, and http://www.clinicaltrials.gov, and screened studies from database inception until March 2011.
Study selection and data abstraction
The definition of ‘inadequate response’ to previous TNFi was based on the reported study inclusion criteria. Our analysis sought RCTs of this patient population that compared a new biological treatment (combined with synthetic DMARD) with a placebo arm using synthetic DMARDs only. We included RCTs in adult RA populations, published in full text and in English. Studies had to report on efficacy or safety. Efficacy was defined as rates of American College of Rheumatology (ACR) (20%, 50% and 70%) response, EULAR (The European League Against Rheumatism) response criteria, or achieving remission (or a low disease activity state). Safety outcomes extracted at the study level included any adverse events (AEs), serious AEs (SAEs), serious infections and infusion- or injection-related reactions after a follow-up of ≥8 weeks.
Data extraction and quality assessment
We extracted patient characteristics and disease activity at baseline as well as ACR response rates and AEs at the end of follow-up. We assessed study quality using the Jadad criteria.5
Data synthesis and analysis
Using Cochrane's Q, we tested for heterogeneity between studies (significant if p<0.10) and quantified the extent of variability between studies due to heterogeneity instead of chance with I2 (ranging from 0% to 100%, with a value >50% indicating substantial heterogeneity).6 We tested for clinical diversity by examining and testing baseline study population characteristics for heterogeneity as a prerequisite for pairwise indirect meta-analysis—that is, homogeneity allows comparison of treatment arms across different trials (as opposed to the comparison of treatment arms within the original trials). Because no head-to-head trials directly comparing two new drugs were identified, we used indirect meta-analysis to estimate the relative effectiveness of two active interventions indirectly through a common comparator using the method of Bucher.7 ,8 For all biological agents, we derived the clinical efficacy as the OR of achieving an ACR response after 24 weeks and calculated the risk difference for safety outcomes compared with placebo. In pairwise indirect meta-analyses of drug efficacy (overall response rates) and safety, we compared each biological agent with each of the other biological treatments. We also compared efficacy after one previous TNFi with outcomes after multiple TNFi drug failures. We applied a random effects model, using DerSimonian Laird statistics9 and carried out all analyses with MetaAnalyst β 3.13 software.10 The random effects model accounts for variance between and within studies when combining summary study outcomes (ACR20, 50 and 70).
We report outcomes according to the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) statement.11
Results
Study selection and evaluation
The Medline search yielded 1268 citations, 50 publications for detailed review and four RCTs for final inclusion12,–,15 (figure 1).

Search and selection process. DMARD, disease-modifying antirheumatic drug; FU, follow-up; IR, inadequate response; RA, rheumatoid arthritis; RCT, randomised controlled trial
Studies included in the systematic review
The evaluated treatment options comprised abatacept (ABA), golimumab (GOL), rituximab (RTX) and tocilizumab (TOC). We included the 50 mg arm of GOL and the 8 mg arm of TOC in our primary analyses. Table 1 lists the baseline characteristics and study details.
Patient baseline characteristics
Qualitative synthesis
For inclusion, all four trials required patients to have active disease despite TNFi; but specific criteria differed—for example, minimal swollen joint counts required were ≥4,14 ≥6,13 ≥8,12 or ≥10,15 minimal tender joint counts were ≥4,14 ≥8,12 ,13 or ≥12.15 In addition, three studies12 ,13 ,15 required acute phase reactant elevations for inclusion, defined as either C-reactive protein >1.5 mg/dl12 or >1.0 mg/dl,13 ,15 or erythrocyte sedimentation rate of >28 mm/h.12 ,13 Nevertheless, patient baseline characteristics were similar (table 1), and the tests for heterogeneity proved negative for all investigated baseline parameters: The calculated I2, indicating the proportion of variability in the trials that is attributable to between-study variation, ranged from 0% to 6.2% for baseline characteristics, including measures of disease activity (joint counts, acute phase reactants and functional status), and patient characteristics such as age or disease duration.
Inadequate response (as opposed to safety-related termination) was the reason for discontinuation of previous biological treatments in 58%14 to 95%13 of the enrolled patients. In all trials, the previous TNFi treatments were adalimumab, etanercept or infliximab, but patients could have been treated with more than one of them.
The investigated agents were administered in combination with MTX in all trials. However, in the ATTAIN (Abatacept Trial in Treatment of Anti-TNF INadequate responders) trial,15 patients could receive synthetic DMARDs other than MTX in combination with ABA and in the GO-AFTER (Golimumab in patients with active rheumatoid arthritis after treatment with tumour necrosis factor α inhibitors) study about 30% of the patients received only GOL monotherapy.14 To match the other trials better, we therefore obtained results of the patient subpopulation treated with concomitant MTX for two time points of the GO-AFTER trial.
All trials reported efficacy outcomes in terms of ACR response rates. The ATTAIN-,15 REFLEX (Randomized Evaluation of Long-Term Efficacy of Rituximab),12 and RADIATE (RheumAtoiD arthritIs study In Anti-TNF failurEs) 13 trials defined ACR20 response at week 24 as their primary efficacy outcome, and ACR50 and 70 response rates as secondary outcomes. In the GO-AFTER trial,14 the primary end point was the ACR20 response at week 14. However, to achieve better comparability of follow-up times, we also used results from the assessment at week 24 after GOL start. We assessed these data for our primary analyses, but included the 14-week outcomes in sensitivity analyses.
Additional efficacy outcomes were reported by Emery et al,13 Cohen et al,12 and Smolen et al,14 who described EULAR good and moderate responses at week 24,12,–,14 as well as Disease Activity Score remission and low disease activity,13 ,14 mean change in Disease Activity Score,12 functional outcome using the Health Assessment Questionnaire,12,–,14 and Functional Assessment of Chronic Illness Therapy-Fatigue.12 ,14
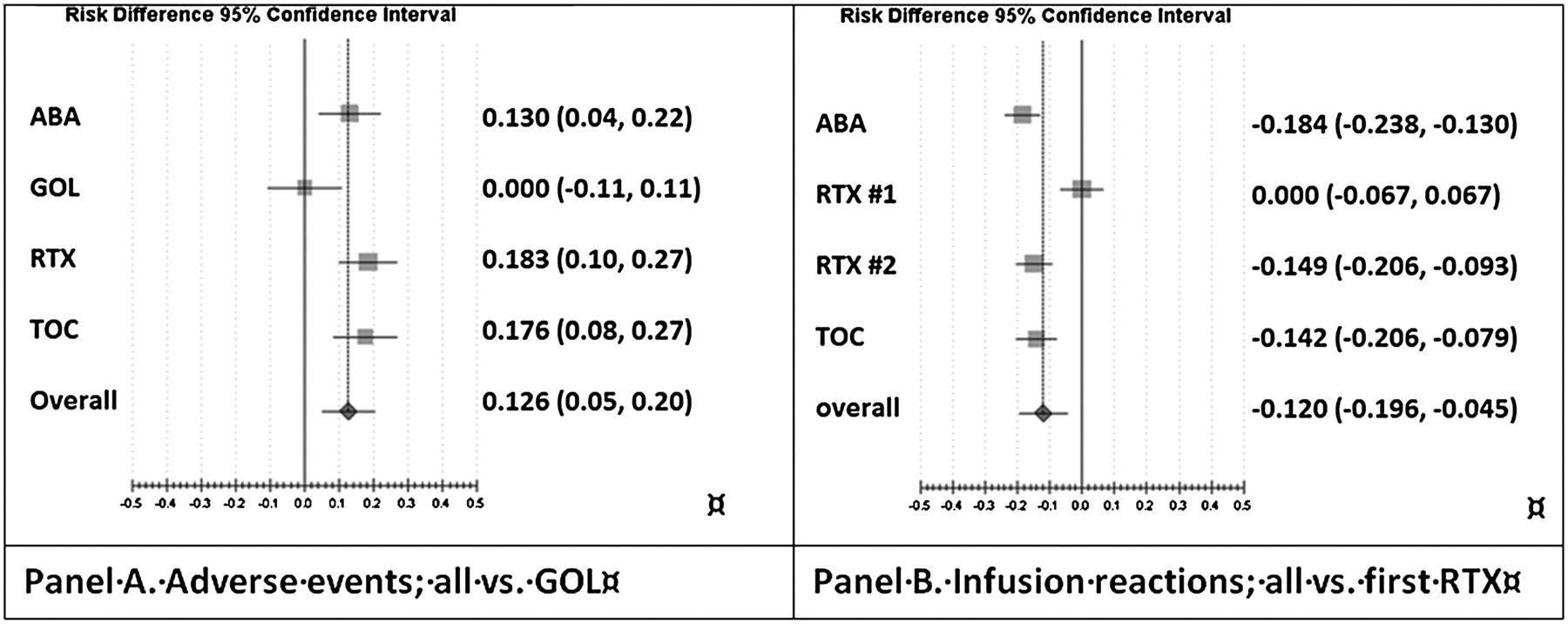
All trials reported AEs, SAE and serious infections. ABA, RTX and TOC trials reported infusion reactions occurring during or within 24 h after the infusion for these three intravenous drugs, and the GOL trial reported injection site reactions for this subcutaneous drug.
We assessed all trials using the five-point Jadad score, that appraises the quality of trial reporting, specifically, a description of randomisation, blinding, withdrawals and dropouts. All included trials were rated between 3 and 5 points, which is usually considered high quality and consistent with equally valid evidence, so no further distinction was applied (table 1).
Quantitative synthesis
Clinical efficacy
Mean pooled ORs for ACR response against placebo were 4.90 (ACR20), 7.20 (ACR50) and 7.43 (ACR70; figure 2). In pairwise indirect comparisons of the biological agents there was no significant efficacy difference in the more profound ACR50 and ACR70 outcomes, which were secondary end points in all studies. ACR20 response rates reported for GOL were lower than for ABA (OR; 95% CI 0.58; 0.36 to 0.92), RTX (0.56; 0.36 to 0.89) and TOC (0.59; 0.36 to 0.96; figure 3); however, note that1 the GO-AFTER study was powered for the 14-week time point and not 6 months,2 30% of GOL patients did not receive MTX and3 some placebo-treated patients received rescue treatment at week 16. Consequently, we performed subanalyses to evaluate the robustness of our results. In those trials that defined the primary end point at week 24, we sought outcomes at earlier time points and compared them with the GOL primary end point at 14 weeks. Detailed comparable data were reported only in the ATTAIN trial: ABA at day 85 did not have a significantly different odds of ACR20 in comparison with GOL (MTX-taking patients, 50 mg and 100 mg combined group) at week 14 (OR=0.78, 95% CI 0.54 to 1.13, p=0.19). Subgroup analyses of the number of previous TNFi failures were available for GOL and TOC: 52 patients who had previously received two TNFi and 26 who had received three TNFi were included in the TOC 8 mg treatment arm. From the GO-AFTER 24 week follow-up, we included data of 22 patients for whom two previous TNFi had failed, and 13 for whom three previous TNFi had failed (notably, the long-term extension data excluded patients who had received rescue treatment after the primary end point of 14 weeks). A comparison of these subgroups with patients who had a history of only one TNFi-IR at inclusion (92 receiving TOC and 68 GOL), showed that ACR20, 50 and 70 response rates did not differ significantly after multiple previous treatments. In indirect comparison of response rates between GOL and TOC, we again found very similar rates after one, two or three TNFi. ACR20 response rates were not different after one, two or three TNFi between GOL and TOC. Although there was a trend toward significance after three TNFi, the small number of patients in this subgroup limits the ability to draw any firm conclusion (figure 4).

Drug efficacy. Direct comparisons of all drugs with their respective disease-modifying drugs and placebo arm; (A) American College of Rheumatology (ACR)20, (B) ACR50, (C) ACR70 abatacept *mixed population of patients receiving methotrexate (MTX; 75%), and patients receiving monotherapy. Golimumab (GOL)**subgroup of patients receiving a combination of GOL and MTX.

Efficacy in pairwise comparisons. similar success rates of abatacept (ABA), golimumab (GOL), rituximab (RTX), tocilizumab (TOC). Top to bottom: ABA is comparator versus GOL, RTX, TOC; GOL versus ABA, RTX, TOC; RTX versus ABA, GOL, TOC; TOC versus ABA, GOL, RTX. OR (95% CI) for American College of Rheumatology (ACR)20 (left column), ACR50 (middle column) and ACR70 (right column) are displayed in red. Black vertical lines indicate an OR of 1. ORs>1 (right of the black line) favour the comparator drug shown in red, ORs<1 (left of the black line) indicate poorer response rates than with the red comparator drug shown in red.

Efficacy after multiple tumour necrosis factor inhibitor (TNFi) failures. Response rates of golimumab (GOL) and tocilizumab (TOC). American College of Rheumatology (ACR)20 (blue lines), ACR50 (green lines) and ACR70 (orange lines) of patients for whom one (top), two (middle) and three (bottom) TNFi had previously failed.
Drug safety
Pooled risk ratios for the occurrence of any AEs were 1.0 (95% CI 0.9 to 1.1), indicating no higher risk in treatment groups in direct comparisons with their respective placebo arms. Similarly, the pooled risk ratios for SAEs, (RR; 95% CI 0.9; 0.6 to 1.2) and serious infection rates (1.3; 0.7 to 2.5) were not significantly different from placebo.
In indirect pairwise comparisons, GOL had significantly fewer general AEs, risk differences (RDs) ranged from 0.13 versus ABA to 0.18 versus RTX and TOC (figure 5). Acute (ie, occurring within 24 h) reactions to infusions were raised in RTX in comparison with ABA (RD 0.18; 0.13–0.24) and TOC (RD 0.14; 0.08–0.21). The second RTX infusion, however, showed no higher risk (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Drug safety. (A) Pairwise indirect comparison against golimumab (GOL): abatacept (ABA), rituximab (RTX) and tocilizumab (TOC) show higher general adverse event rates. (B) Infusion reactions within 24 h: pairwise indirect comparison against the first RTX infusion (RTX #1) shows higher risk than in TOC and ABA.
Discussion
We assessed the comparative effectiveness and safety of the four biological treatment options available for patients with RA when confronted with TNFi non-response. Despite differences in inclusion criteria, testing for heterogeneity in the baseline characteristics was negative, supporting the comparability of the trial populations and allowing for pairwise indirect meta-analysis using the respective DMARD/placebo comparator arm. In the absence of head-to-head trials, our analysis finds that ACR20 response rates at 24 weeks were similar for ABA, RTX and TOC and appeared lower for GOL. The primary outcome of the golimumab GO-AFTER trial, however, was defined at 14 weeks and not 24 weeks, so placebo patients not reaching the primary outcome at week 16 received rescue treatment. The similarity in the more profound ACR50 and 70 response rates for all agents, ABA, GOL, RTX and TOC suggests that all biological drugs have comparable efficacy in TNFi-IR patients with RA.
This conclusion is also supported by the sensitivity analyses comparing data on ABA at day 85 with those of GOL (50 mg and 100 mg groups combined) at week 14 which did not show significant differences, in contrast to the main analysis. Moreover, ACR response rates did not differ significantly when assessing separately patients for whom two or even three TNFi had failed. However, failure of three previous TNFi occurred in relatively few patients and, in general, data were sparse with large CIs. Further data would enhance the accuracy and precision of these comparisons.
Pooled risk ratios of AEs in all trials did not differ significantly between the respective biological agents and their control groups. The observation of lower rates of AEs for GOL might be chance, although it might also be a consequence of ‘selecting-out’ patients susceptible to AEs of TNF inhibition by the previous TNFi treatments.
Adequately powered direct head-to-head comparisons of these biological agents would provide the least biased estimates of efficacy, but such a trial would require a large sample size given the relatively similar efficacy of these agents and be quite expensive. In the absence of such a trial, our study enables an indirect comparison of biological agents with each other and thereby provides some clinical guidance based on best available evidence. Nonetheless, in a validation study, Song et al found a significant discrepancy between direct and adjusted indirect meta-analysis in three of 44 studies.16 Known methodological flaws in indirect meta-analysis include ‘inappropriate search and selection of relevant trials, use of inappropriate or flawed methods, lack of objective and validated methods to assess or improve trial similarity, and inadequate comparison or inappropriate combination of direct and indirect evidence.’16
A recently published comparison of biological agents in MTX- and TNFi non-responders found equal ACR50 rates,17 yet did not analyse ACR70 rates, or safety outcomes. In clinical practice, TNFi switching often occurs before changing to one of the newer non-TNFi biological agents. A meta-analysis of observational studies supports the benefit trying a second TNFi.18 Others suggest that RTX has better efficacy than a second TNFi (except for GOL).19 ,20 Apart from the GO-AFTER study, however, no randomised trials are available, so we could not include any other TNFi other than GOL in our analysis.
Our extensive literature search ensures the inclusion of all published data in TNFi non-responders. We considered the comparability of data and provided sensitivity analyses based on alternative time points to evaluate the robustness of our results, and the absence of direct evidence precluded their incorporation. We provide analyses of relevant efficacy outcomes ACR50 and 70, and also considered drug safety. In addition, our subanalyses give information on success rates after multiple TNFi failure.
The primary limitation of our study is the paucity of RCTs and the absence of head-to-head trials of biological agents. Second, the absence of individual patient level data allows us to analyse only group-level outcomes. Third, only two of the four studies in our analysis report on incomplete response to TNFi (58% in GO-AFTER and 95% in RADIATE). Insufficient response and other reasons for termination of previous TNFi treatment are not usually distinguished in the results of RCTs which is necessarily reflected in our analysis. This common practice highlights the need for future trials to report results separately for patients with inadequate response from those with other reasons for termination. A further limitation—namely, uncertainty about the comparison of the ACR response rates for GOL with the other agents has been detailed above.
In conclusion, in this patient group characterised by disease refractory to multiple previous treatments, significant improvement is possible with approved biological agents, which also show acceptable safety outcomes in the studied trial populations. Thus, until additional studies become available, other criteria besides safety and efficacy, may play a role in therapeutic decision-making, including costs, patient preferences about route of administration and clinician familiarity with the treatment. Nonetheless, our findings should reassure both clinicians and patients that each of the available agents may be beneficial.
References
Footnotes
Funding This study was supported by a grant from Schering-Plough.
Competing interests Disclosure of honoraria for advice or public speaking/grants received/advisory board of the authors are as follows: MS: Schering Plough, Pfizer; DA: Pfizer, MSD, UCB, Roche, Schering Plough; JS: Abbott, BMS, Celgene, Centocor, MSD, Novo, Pfizer, Roche, UCB, BMS, Royalties: Elsevier.
Provenance and peer review Not commissioned; externally peer reviewed.